Article Text

Statistics from Altmetric.com
- ACGII/HCG, achondrogenesis II/hypochondrogenesis
- COL2A1, type II collagen gene
- FGFR3, fibroblast growth factor receptor 3 gene
- PLSD, platyspondylic lethal skeletal dysplasia
- PLSD-L, platyspondylic lethal skeletal dysplasia, Luton type
- PLSD-SD, platyspondylic lethal skeletal dysplasia, San Diego type
- PLSD-T, platyspondylic lethal skeletal dysplasia, Torrance type
- TD, thanatophoric dysplasia
- TD1, TD2, two types of thanatophoric dysplasia
- SEDC, spondylo-epiphyseal dysplasia congenita
Platyspondylic lethal skeletal dysplasias (PLSDs) are a heterogeneous group of chondrodysplasias characterised by severe platyspondyly and limb shortening. The most common form of PLSD is thanatophoric dysplasia (TD), which has been divided into two types, TD1 (MIM 187600) and TD2 (MIM 187610). Three other types of PLSD, or TD variants have been distinguished from TD, the San Diego (PLSD-SD; MIM 270230), Torrance (PLSD-T; MIM 151210), and Luton (PLSD-L; MIM 151210) types.1,2 PLSD-L is now considered to be a mild phenotypic variant of PLSD-T. Mutations in the fibroblast growth factor receptor 3 (FGFR3) gene were identified in TD and PLSD-SD,3 but not in PLSD-T and PLSD-L.3,4
PLSD-T is a common subtype of PLSD.2 The radiological characteristics include wafer-like vertebral bodies, severe hypoplasia of the lower ilia, extremely short long bones with ragged metaphyses, and bowing of the radius. Its chondro-osseous histology is characterised by hypercellularity with slightly large chondrocytes in the resting cartilage and normal columnisation with incorporation of cartilage into bone at the chondro-osseous junction.1,2 These radiological and histological findings of the disorder can be used to discriminate it from other lethal or semilethal skeletal dysplasias including TD.
Perinatal death is generally considered to be inevitable in PLSD-T. Recently, however, non-lethal phenotypes of the disorder with better ossified vertebral bodies have been proposed, based on the observations of two affected families. One family included an affected mother who survived to adulthood and her affected daughter who died soon after birth,5,6 and the other family included an affected mother and her son, both of whom are living.4 These observations raise the question of whether PLSD-T represents a single entity with a wide clinical spectrum or a heterogeneous group of disorders with superficial radiological similarities. Here we describe two examples of the disorder: one perinatally lethal case and one mild, surviving case. In both, we identified causal mutations in the C-propeptide region of the type II collagen gene (COL2A1).
SUBJECTS, METHODS AND RESULTS
Clinical reports
Patient 1
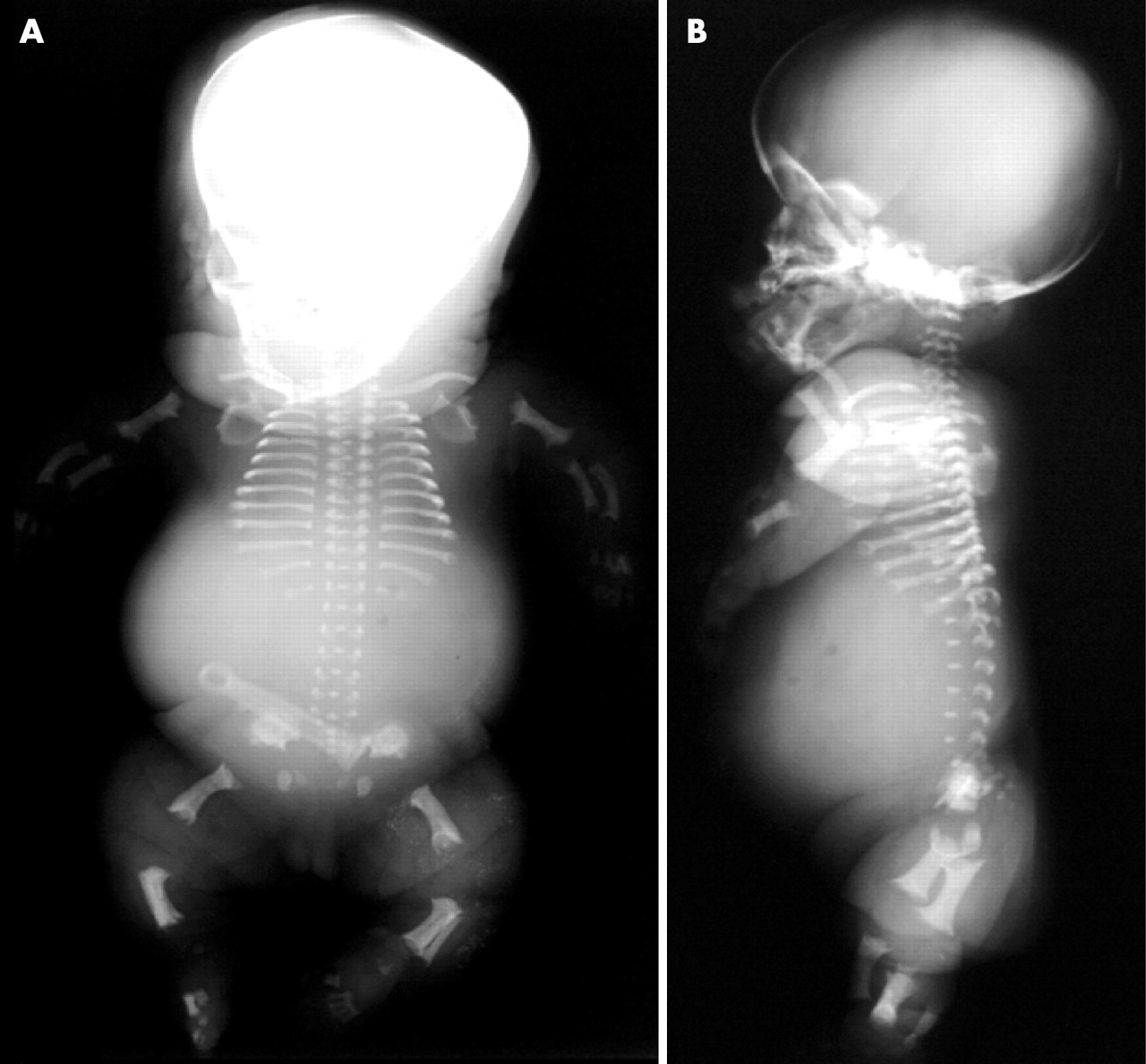
Patient 1, an affected girl, was the first child of non-consanguineous parents, both of whom had hearing impairment. Prenatal ultrasonography at 29 weeks of gestation revealed polyhydramnios, thoracic hypoplasia and micromelia. Femoral length was 26.6 mm (the mean of 16 weeks of gestation), and biparietal diameter was 78.3 mm (the mean of 31 weeks of gestation). She was delivered and stillborn at 34 weeks of gestation. Body length was 31.0 cm, weight was 2090 g, occipital frontal circumference was 34.0 cm, arm length was 9 cm, and leg length was 10 cm. A flat face with a depressed nasal bridge, low set ears, thoracic hypoplasia, and a protuberant abdomen were noted. Postmortem radiographs showed a hypoplastic thorax, wafer-like vertebral bodies, craniocaudal shortening of the lower ilia with medial bony projections, and extreme shortening of the long bones with cupped, ragged metaphyses (fig 1). The radius was bowed. An autopsy revealed no visceral malformations other than pulmonary hypoplasia.

Postmortem frontal (a) and lateral (b) radiographs of patient 1, with the lethal phenotype of platyspondylic skeletal dysplasia, Torrance type. Note the wafer-like vertebral bodies, severe iliac hypoplasia with medial bony projections, shortening of the long bones with ragged metaphyses, and bowing of the radius.
Key points
-
Platyspondylic lethal skeletal dysplasia, Torrance type (PLSD-T) is a severe form of chondrodysplasia that belongs to the group of platyspondylic lethal skeletal dysplasias. Its aetiology remains to be determined. While most reports of PLSD-T describe sporadic and perinatally lethal cases, the presence of cases having longer survival times and dominant inheritance have been suggested recently.
-
We describe two sporadic PLSD-T patients; the first was perinatally lethal, while the second is living after surviving respiratory failure in infancy. The stillborn patient showed the radiological hallmarks of the disorder, including wafer thin vertebral bodies, severe hypoplasia of the lower ilia, and extreme shortening of the long bones with ragged metaphyses. Our histological findings for this patient were also quite consistent with PLSD-T. In the living patient, radiological manifestations during the neonatal period were identical to those of the stillborn patient, except that the vertebral bodies were ossified better. In this patient, the neonatal PLSD-T phenotype evolved into that of Kniest-like dysplasia in childhood.
-
In both patients, molecular analysis has revealed de novo heterozygous mutations in the C-propeptide region of the type II collagen gene (COL2A1).
-
Our results expand the number of phenotypes associated with COL2A1 mutations and reclassify PLSD-T as a variant of type II collagenopathies that has a wide clinical spectrum. Milder phenotypes of PLSD-T may be misdiagnosed as other skeletal dysplasias due to age dependent phenotypic evolution.
Patient 2
Patient 2, an affected boy, was born to healthy, non-consanguineous parents. He was vaginally delivered at full term. His birth length was 39.0 cm (−5.9 SD), weight was 3140 g (−0.2 SD), and occipital frontal circumference was 36.0 cm (+1.8 SD). Mid-face hypoplasia, a hypoplastic thorax, and rhizomelic shortening of the limbs were noted. Soon after birth, he showed respiratory distress. Respiratory support with intratracheal intubation was continued from 2 days to 6 months of age. Gross motor development was retarded: head control was observed at 7 months, crawling and standing with support at 15 months, and walking alone at 25 months of age. Mental development was initially retarded but reached the normal range with advancing age. At 11 months of age, he was referred to us for further evaluation. At 11 months, his body length was 53.2 cm (−8.6 SD), weight was 5495 g (−3.8 SD), occipital frontal circumference was 45.6 cm (the mean), and chest circumference was 35.0 cm (−5.6 SD). Laboratory examinations, chromosome analysis, and brain computed tomography all gave normal results. Patient 2 received growth hormone therapy from 3 1/2 years to 5 years of age without acceleration of growth rate. At 5 years of age, neither ophthalmological abnormality nor hearing impairment had developed. Skeletal surveys were performed in the neonatal period (fig 2), and at 2 and 5 years of age (fig 3). In the neonatal period, the radiological manifestations included a hypoplastic thorax, ovoid vertebral bodies, hypoplasia of the lower ilia, and extreme shortening of the long bones with cupped, ragged metaphyses. With advancing age, platyspondyly became less conspicuous along with the development of thoracolumbar gibbus, the long bones showed dumbbell deformity and fluffy mega-epiphyses, resembling those of Kniest dysplasia, and brachydactyly became manifest with accelerated epiphyseal maturation of the short tubular bones. The radiological manifestations at 2 and 5 years were essentially the same.

Frontal (a) and lateral (b) babygrams of patient 2, with the mild phenotype of platyspondylic skeletal dysplasia, Torrance type in the neonatal period. Note the ovoid vertebral bodies, iliac hypoplasia, and shortening of the long bones with ragged metaphyses.

Radiographs of lateral spine (a) and lower legs (b) in patient 2 at 5 years of age. Platyspondyly becomes less conspicuous along with development of thoracolumbar gibbus. Dumbbell deformity of the long bones is noted.
Histological examination
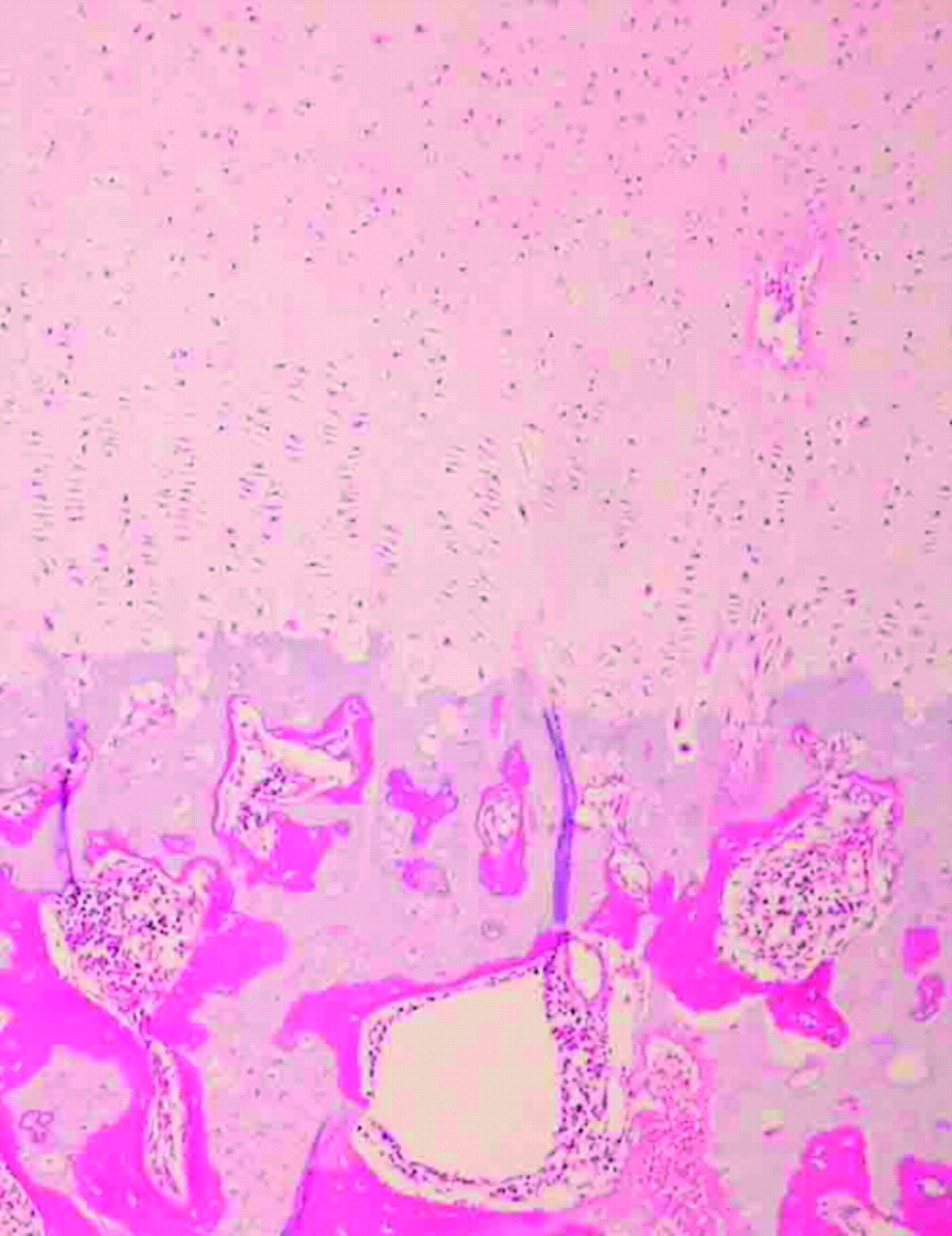
Samples of the distal femur and lumbar vertebral column were dissected from patient 1 and processed for regular microscopic examinations. Histological findings from these samples were similar, but there were some notable differences (figures 4 and 5). In the distal femur, the epiphyseal (resting) cartilage consisted of an admixture of hypercellular and hypocellular areas. Resting chondrocytes were slightly enlarged, and some were vacuolated. The hypocellular zones showed matrix degeneration and contained dead chondrocytes. Vascular canals in the epiphyseal cartilage were not increased in number. Columnar formation at the chondro-osseous junction was defined but incomplete. Hypertrophic cartilage with a columnar arrangement was incorporated into metaphyseal bone trabeculae. Differences in the vertebra included less conspicuous hypocellular zones and relatively well preserved columnar formation. Bone trabeculae were irregularly intermingled with cartilage.

Histology of the distal femur (high power views, haematoxylin-eosin staining): (a) epiphyseal cartilage, and (b) chondro-osseous junction. Note the admixture of hypercellular areas and hypocellular areas, slightly enlarged chondrocytes, and incomplete columnar formation.

Histology of the vertebral body (low power view, haematoxylin-eosin staining). Note the relatively well preserved columnar formation and irregular bone trabeculae intermingled with cartilage.
Molecular analysis
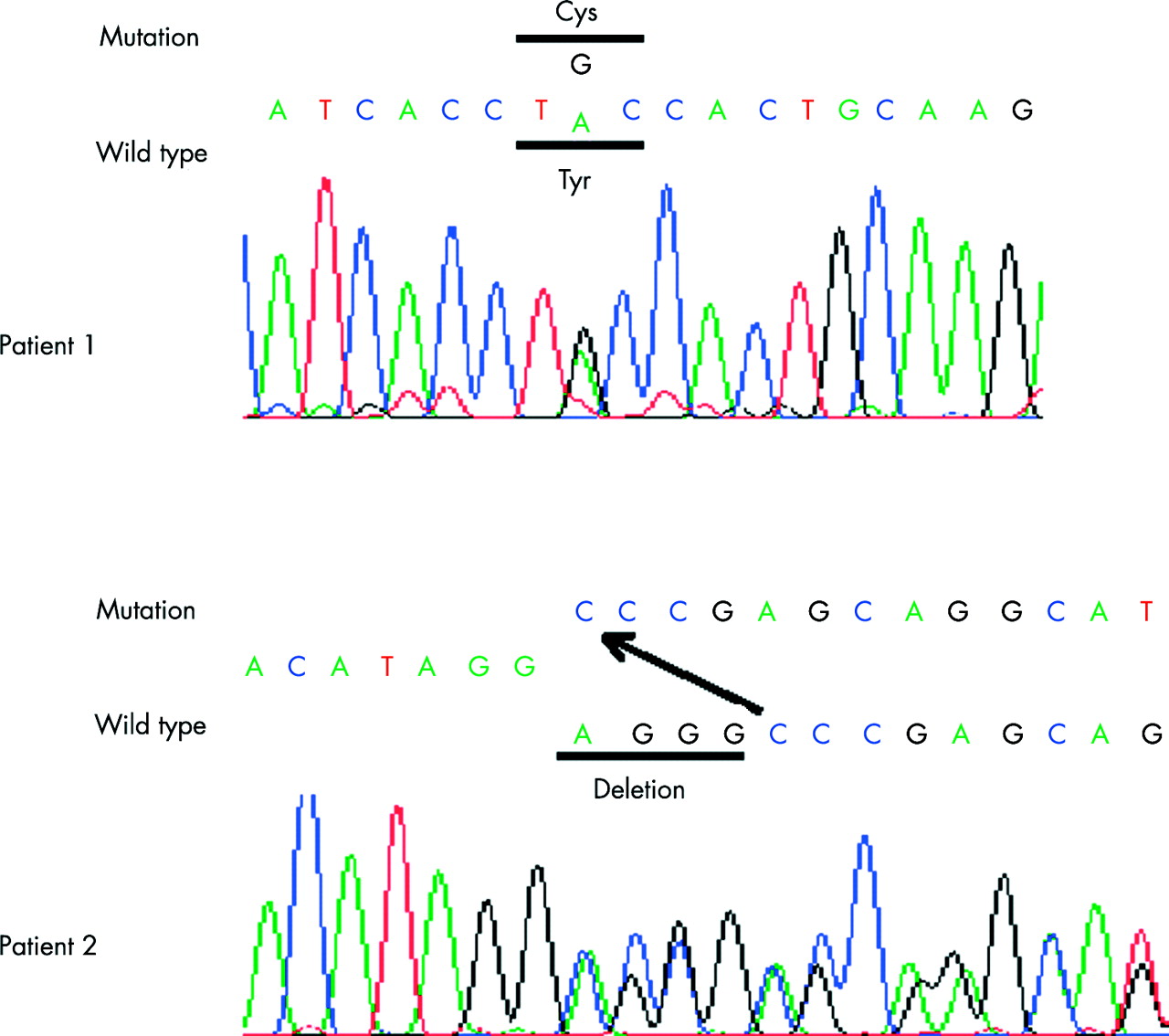
Blood samples were obtained from patients and their parents with informed consent, and genomic DNA was extracted using standard procedures. Coding regions of the entire COL2A1 gene, along with flanking intronic regions, were amplified by PCR using previously described primer sets.7 PCR was performed with the exTaq system (Takara Shuzo, Otsu, Japan) according to the manufacturer’s instructions. PCR products were directly sequenced using an ABI Prism 3700 automated sequencer and the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster, CA). In patient 1, we identified a missense mutation, c.4172A>G (translation start site of NM_001844 is designated as +1.) in exon 53, which predicts a tyrosine to cysteine substitution (Tyr1391Cys; amino acid (aa.) 177 of the C-propeptide). In patient 2, we identified a 4-bp deletion, c.4413-6del4 in exon 54, which predicts early termination at codon 1480 (aa. 266 of the C-propeptide) (fig 6). Both mutations are de novo and were not found in 100 control subjects.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
COL2A1 mutations in the C-propeptide region in patients with platyspondylic skeletal dysplasia, Torrance type. Top (patient 1): A missense mutation, c.4172A>G at codon 1391 (codon 177 of the C-propeptide) that results in a tyrosine (Tyr) to cysteine (Cys) substitution. Bottom (patient 2): A 4-bp deletion (AGGG) that results in a frameshift and an early termination codon (p.Gly1472fsX8).
DISCUSSION
Patient 1 is considered to represent the lethal PLSD-T phenotype. The PLSD-T phenotype shares many radiological features with the achondrogenesis II/hypochondrogenesis (ACGII/HCG) spectrum (MIM 200610). However, the radiological manifestations observed in patient 1 are distinct from those common to the ACGII/HCG spectrum. Specifically, the severe hypoplasia of the lower ilia, the ragged metaphyseal appearance, and the bowing of the radius observed in patient 1 warranted a diagnosis of PLSD-T. The histological manifestations in patient 1 were identical to those of a previously reported case of PLSD-T.8 While histological findings in PLSD-T can vary between cases,1,9 several characteristics distinguish this disorder from the ACGII/HCG spectrum. These include the absence of increased number of vascular canals in cartilage, less conspicuous hypercellularity, and a relatively preserved columnar formation, all of which were observed in patient 1.
The neonatal phenotype of patient 2 was consistent with mild PLSD-T phenotypes with better ossified vertebral bodies previously described.4–6 The phenotype of the patient showed metatropism, or age-dependent evolution, wherein the initial PLSD-T phenotype evolved into Kniest-like dysplasia. These observations are compatible with a previously described patient with non-lethal PLSD-T, who had severe metaphyseal flaring in childhood,4 but they contrast with other patients with non-lethal PLSD-T, whose later symptoms were classified as Stickler-like dysplasia and an unclassifiable spondyloepiphyseal dysplasia.4,5 The later phenotypic differences between these patients can be regarded as variations of similar conditions, because from a pathogenic viewpoint, Stickler dysplasia (MIM 108300) and Kniest dysplasia (MIM 156550) are considered closely related.10 Unclassifiable spondyloepiphyseal dysplasias also constitute a part of type II collagenopathy.
A type II collagen defect has been suspected in PLSD-T based on protein analysis of cartilage matrices; however, this defect has previously been unconfirmed on a molecular level.11COL2A1 mutations create a multiplicity of clinical entities of varying severity; these are collectively described as type II collagenopathies.10 A large group of type II collagenopathies results from mutations in the triple helical region of the protein, and a small group correlates with mutations in the C-propeptide region. The former group presents as the ACGII/HCG spectrum, spondylo-epiphyseal dysplasia congenita (SEDC (MIM 183900)), Stickler dysplasia, and Kniest dysplasia. The latter group comprises very few cases, which were reported to manifest as a variety of clinical phenotypes, including SEDC,12 hypochondrogenesis,13 spondyloperipheral dysplasia,14 and distinctive phalangeal epiphyseal dysplasia.15 Our molecular analysis shows that PLSD-T also belongs to this latter group.
Genotype-phenotype correlations for C-propeptide mutations in COL2A1 remain elusive. Unlike the mutation identified in patient 1, previously reported missense mutations in the C-propeptide region predict the substitution of non-cystine amino acids. This may explain some observed phenotypic differences. In addition, one previously reported case, caused by an exon 51 mutation that led to a premature termination codon, showed spondyloperipheral dysplasia, characterised by a combination of SEDC-like manifestations with type E-like brachydactyly resulting from premature epiphyseal maturation of the short tubular bones.14 It is intriguing that patient 2, whose COL2A1 mutation also predicts premature termination, shows similar accelerated epiphyseal maturation of the short tubular bones.
It is also intriguing that patient 1 showed medial bony projections of the ilia, which resembled those of Schneckenbecken dysplasia (MIM 269250).2 Similar findings are evident in the illustrations of the previous reports, although those did not attract particular attention.2,4 Schneckenbecken dysplasia may also be caused by COL2A1 mutations.
Our identification of COL2A1 mutations in both lethal and non-lethal phenotypes of PLSD-T indicates that the disorder represents a single entity inherited as an autosomal dominant trait, rather than a heterogeneous group of disorders with superficial radiological similarities. The phenotypic variation may be significant. Mildly affected individuals may be misdiagnosed as other type II collagenopathies because of the phenotypic metatropism. Examination of COL2A1 would aid in the correct diagnosis of those cases. The differential diagnosis between mild PLSD-T and other type II collagenopathies is beneficial, because vitreoretinal degeneration and hearing impairment, which are common syndromic constituents in other type II collagenopathies, do not seem to occur in mild PLSD-T.
REFERENCES
Footnotes
-
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports and Science of Japan (contract grant number: 14370476).