Article Text

Statistics from Altmetric.com
Retinitis pigmentosa (RP), which occurs in about one in 3000–7000 people in Spain, is inherited in an autosomal dominant manner in 12% of cases, in an autosomal recessive way in 39%, and in an X linked manner in 4% of cases. This leaves 41% of RP cases with a simplex form and 4% in which the transmission pattern is unclear.1
Direct analyses of rhodopsin, the alpha and gamma subunits of rod cGMP-phosphodiesterase, periferin/RDS, rod outer segment membrane protein, recoverin, guanilate cyclase activating protein, S antigen, interstitial retinol binding protein, and NRL have failed to detect any disease causing mutation in non-syndromic ARRP Spanish families. Mutations in the beta subunit of the rod cGMP-phosphodiesterase gene,2–5 in the ATP binding cassette receptor gene,6 and in the TULP1 gene7 account for a small percentage of Spanish ARRP families. These data indicate that genes other than these may be involved in the remaining families, emphasising the genetic heterogeneity of the disease and reinforcing the hypothesis that in ARRP a number of genes rather than one major gene will account individually for a small number of cases.
The recent report that a missense mutation in the USH2A gene (C759F) is present in 4.5% of patients with non-syndromic ARRP8 prompted us to analyse the involvement of this mutation in a large set of Spanish ARRP families. A complete mutational analysis of the coding region of the USH2A gene was performed in all cases in which the C759F allele was found. Additional mutations were identified in the USH2A gene in non-syndromic ARRP patients. Interestingly, two C759F homozygotes belonging to a consanguineous ARRP family had no RP symptoms and no hearing impairment.
PATIENTS AND METHODS
A group of 196 unrelated ARRP patients plus four cases of retinitis punctata albescens were studied. The patients were diagnosed at the Hospital de la Santa Creu I Sant Pau (Barcelona) at the Fundación Jiménez Diaz (Madrid) or at the Hospital Virgen del Rocio (Sevilla). The clinical diagnosis of RP was based on ophthalmological examination including measurements of visual acuity, ophthalmoscopy, dark adaptation, perimetry, and electroretinogram amplitudes.
One hundred blood donors (200 chromosomes) were used as controls to evaluate the frequency of the mutations and polymorphisms found in the patient sample. Blood samples were obtained after informed consent was given and genomic DNA extracted from these blood samples was used to assess the involvement of the C759F mutation in the USH2A gene. Intronic PCR primers flanking exon 139 were used for PCR amplification and direct sequencing. All PCR reactions were carried out on a PTC-200 Peltier Thermal Cycler. The PCR products were sequenced using the Thermosequenase Primer Cycle sequencing Kit 7-deaza dGTP (Amersham Pharmacia Biotech UK limited, Buckinghamshire, UK). The sequences were analysed on an automated sequencer Li-Cor DNA Analyser Gene Reader 4200 (Li-Cor Inc, Lincoln, NE, USA).
Key points
-
The USH2A gene has been shown to be associated with Usher syndrome type II and recent data indicate that mutations in this gene could also cause non-syndromic autosomal recessive retinitis pigmentosa (ARRP). We have screened our panel of 196 unrelated ARRP patients for the presence of the C759F missense mutation in the USH2A gene.
-
We have identified compound heterozygotes with C759F and nonsense, splicing, or missense mutations in the USH2A gene that manifest as recessive retinitis pigmentosa without hearing loss. We found four C759F homozygotes belonging to two different consanguineous families; two of them were non-syndromic RP affected patients while the remaining two had no RP symptoms and no hearing impairment.
-
When considering the information presented here together with that in previous reports, a picture emerges of considerable phenotypic heterogeneity arising from mutations in USH2A which ranges from distinct types of Usher syndrome, through non-syndromic retinitis pigmentosa, to unaffected subjects. The wide range of phenotypes associated with usherin mutations underlines how the relationship between pathogenetic mutations and disease phenotype is becoming increasingly complex.
In all cases in which a C759F allele was found, a mutational analysis of the USH2A gene coding region was performed. Primers were designed from the consensus intronic sequences (GenBank accession numbers AF091973-AF091889) of the USH2A gene as described previously.9 PCR products were sequenced as described above.
To establish the haplotype for the C759F alleles, six single nucleotide polymorphisms within the USH2A gene, exons 2–21, were selected as previously described.10 Haplotype analysis was performed on DNA samples from all members of the six families depicted in fig 1. The establishment of haplotypes enabled us to exclude the existence of large genomic deletions in the USH2A gene in the affected RP patients in these families.
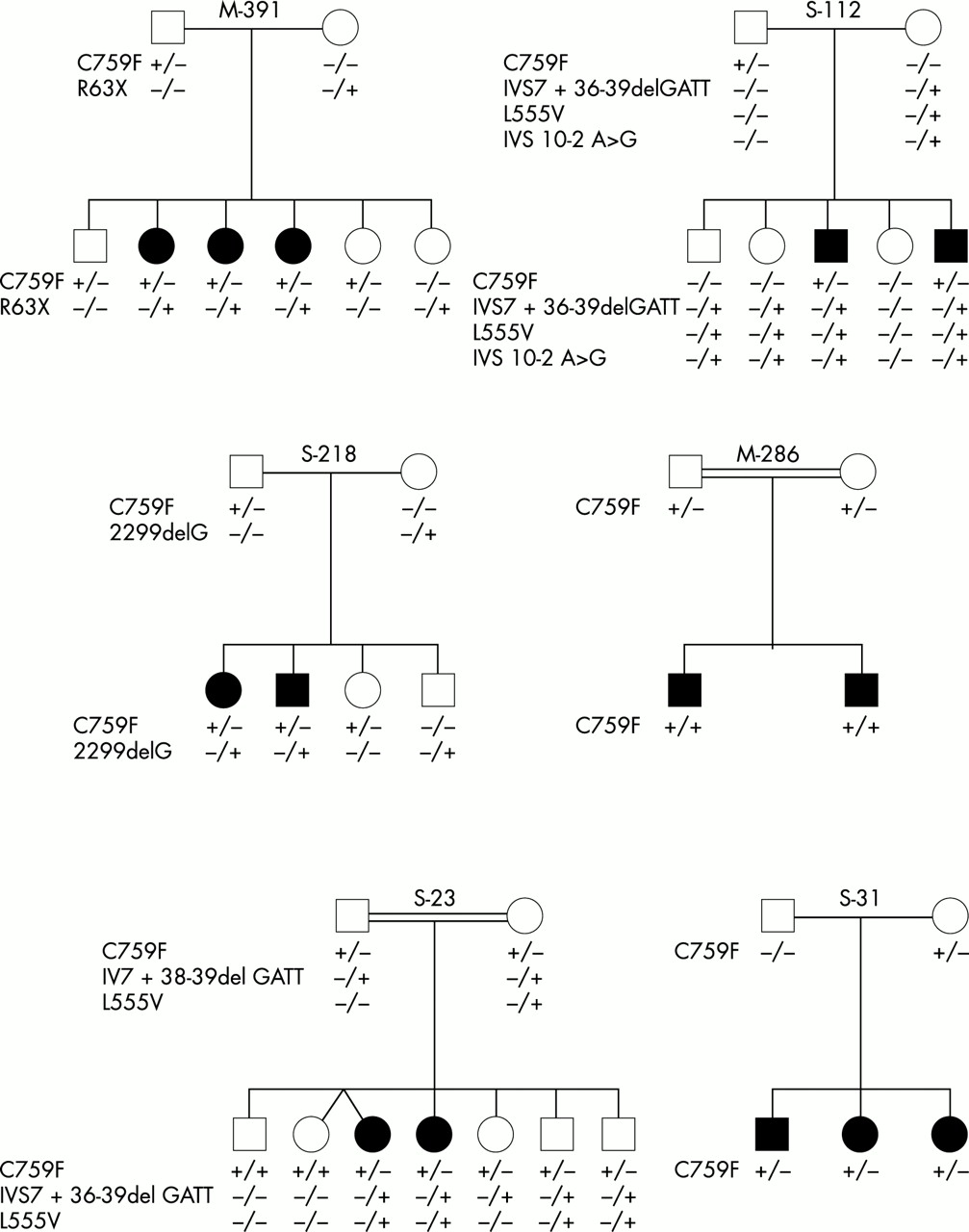
Pedigrees of ARRP families with mutations in the USH2A gene.
RESULTS
In the group of 196 ARRP patients analysed, we identified one homozygote and eight carriers of the C759F (TGC-TTC) missense mutation.
In three out of eight heterozygotes, no family members were available for study. In the remaining cases (five carriers and one homozygous patient), the screening for this mutation was extended to other family members and we identified 21 additional carriers (seven were RP affected patients and 14 were unaffected members of the families) and three additional homozygous cases: one case was the ARRP affected brother of the previous homozygote (M-286) and in another family (S-23) two were unaffected subjects (fig 1).
A complete mutation study of the coding regions of the USH2A gene was performed in (1) one ARRP patient from each of the six families depicted in fig 1, (2) two unaffected ARRP subjects homozygous for C759F from family S-23, and (3) the three ARRP patients who were heterozygous for C759F with no family members available for study.
In family M-391, in which six subjects were carriers of the C759F mutation, we identified the second mutated allele in the three ARRP affected daughters. The C to T transition at nucleotide 187 leads to a premature stop codon at position 63 (R63X). One healthy sister was heterozygous for this mutation and the two remaining unaffected sibs were heterozygous for C759F. In order to assess hearing status, the ARRP patients underwent pure tone audiometry which showed a hearing acuity within the normal range.
In family S-112, the father and the two affected RP brothers were heterozygous for C759F. We identified three variations in one maternal allele: a previously described polymorphism (IVS7 + 36–39del gatt), a replacement of a leucine residue by a valine in codon 555 owing to a C to G transversion at position 1663 in exon 10 (L555V), and an IVS10 −2 A>G change affecting the acceptor splice site of intron 10. This maternal chromosome was inherited along with the C759F mutation in the two affected RP patients. Two unaffected sibs carried the maternal mutant allele only, whereas the remaining healthy sister had no mutations in the USH2A gene.
In family S-218, the affected RP sibs were compound heterozygotes. They had inherited a paternal C759F allele and a maternal 2299delG mutated allele. Two unaffected brothers were heterozygous for these mutations, respectively.
C759F was found in a homozygous state in two affected RP members (aged 50 and 54) of a consanguineous family (M-286). Upon clinical examination and pure tone audiometry, they were found to have hearing acuity within the normal range for their age.
S-23 is also a consanguineous family with two RP affected females aged 44 and 41 years. These patients and two asymptomatic brothers, aged 35 and 27 years, showed an identical pattern: the C759F paternal allele and the maternal allele containing the missense mutation (L555V) and the polymorphism (IVS7 + 36–39del gatt) identified in family S-112. A healthy C759F heterozygous female aged 39 also carried the intron 7 polymorphism, whereas two other unaffected sibs, aged 45 and 44 years, in whom neither L555V nor the intron 7 polymorphism was detected, were C759F homozygotes. A thorough otological and ophthalmological investigation of these two subjects ruled out RP symptoms and hearing impairment. We resampled and reanalysed all the members of this family to exclude the possibility of sample mix up.
In family S-31 in whom the three RP affected sibs were heterozygous for the C759F mutation, the analysis of the coding regions of the USH2A gene failed to detect any additional mutation.
The mutational analysis of the USH2A gene in the three ARRP patients, who were carriers of the C759F mutation and had no family members available for study, allowed the identification of the second mutated allele in one patient, a replacement of a histidine residue by a proline (codon 610) owing to a A to C transversion at position 1829 in exon 10.
During the course of the study, a novel missense mutation in exon 13, P761R, was identified in an ARRP patient. Mutational analysis of the remaining exons failed to detect any other changes.
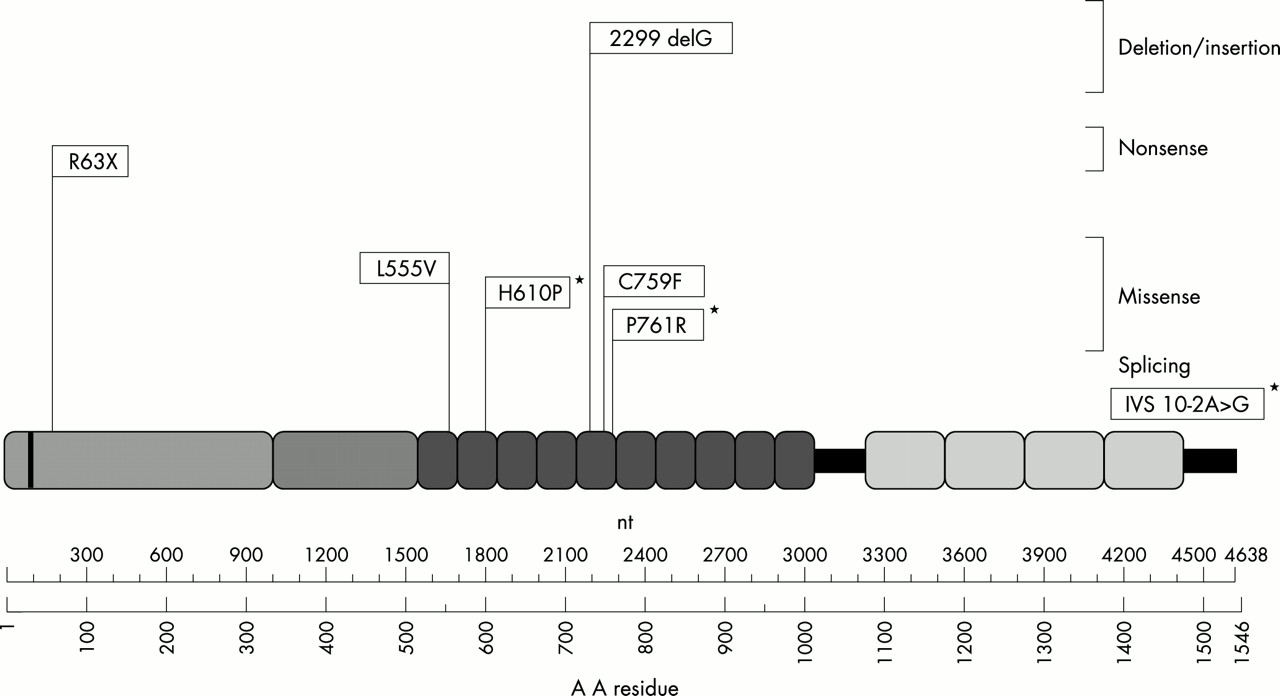
The locations within the usherin protein of the identified mutations in our ARRP patients are indicated in fig 2.
{kind=link}
{kind=link}
Schematic representation of the location within the usherin protein of the mutations identified in Spanish ARRP patients. *Indicates novel mutations.
We identified two alleles with the C759F mutation in a panel of 200 control chromosomes. The missense mutations L555V and P761R were not found whereas a frequent polymorphism (IVS7+36–39 del gatt) was present in 4% of the alleles.
Haplotype analysis within the USH2A gene was performed on all family members in fig 1. Only one haplotype was found to be associated with the C759F mutation. This was the only haplotype found in the four C759F homozygotes, as expected. The remaining affected ARRP patients carried two different haplotypes and were heterozygous for at least two out of six SNPs. Thus, the presence of a large deletion can be excluded.
DISCUSSION
Since the identification of the USH2A gene,11 a number of authors have screened the complete gene in Usher II patients from different ethnic backgrounds.9,12–15 The results obtained illustrate the high frequency of mutations in the USH2A gene in patients meeting the clinical criteria for Usher type II as well as in cases with an atypical USH phenotype.12,13
Recently, Rivolta et al8 indicated that mutations in the USH2A gene could cause a significant proportion of non-syndromic recessive RP given that the C759F allele by itself accounts for approximately 4.5% of the 224 ARRP cases included in the study. We screened our panel of 196 unrelated ARRP patients for the presence of the C759F allele and we found the mutation in nine cases. The frequency of C759F in Spanish patients (4.6%) is thus similar to that reported in patients from North America.8
When investigating the second mutated allele in carriers of C759F, six additional mutations were identified, three of which have not been reported previously (fig 2). The mutations that led directly or indirectly to premature termination of translation, resulting in the disruption of the function of the USH2A protein, were (1) the nonsense mutation R63X, which occurs at a CpG dinucleotide, (2) the deletion 2299delG, which most commonly causes Usher syndrome type IIa, and (3) a novel splicing mutation (IVS10 −2 A>G), which alters the acceptor splice site of intron 10.
The missense mutations identified, two previously described (L555V and C759F) and two novel (H610P and P761R), affect the laminin type epidermal growth factor-like (LE) domains of the protein. LE domains have been shown to be folded protein modules containing looped structures stabilised by disulphide bridges. Substitutions of cysteine residues involved in these structures such as C759F will result in abnormal folding of the LE domain, affecting the properties of the protein. As for the H610 P mutation in exon 10, the replacement of a basic residue by a neutral and hydrophobic amino acid probably has an effect on the function of usherin. A similar conclusion can be drawn for the P761R mutation affecting the same LE domain as C759F, where a conserved and neutral residue is replaced by a basic amino acid. The L555V missense mutation replaces a leucine by a valine residue; both residues are neutral amino acids located in the first LE domain and the effect, if any, of this substitution on the structure and/or function of the protein remains to be explored.
Rivolta et al8 identified two non-syndromic ARRP patients who were compound heterozygotes with C759F and previously reported frameshift mutations, which indicates that the frameshifts do not cause Usher type II, but only non-syndromic RP if they are inherited together with the missense change C759F. We can confirm this assumption because in Spanish patients additional compound heterozygotes with C759F and nonsense, splicing, or missense mutations are associated with identical phenotypic features, reinforcing the hypothesis that mutations in the UHS2A gene can result in ARRP without hearing loss.
The four C759F homozygotes found in the present study belonged to two families, M-286 and S-23 (fig 1). M-286 was a consanguineous family in which the two RP sibs were homozygotes for the C759F mutation. Upon clinical examination and pure tone audiometry, they were found to have hearing acuity within the normal range for age. More interestingly, S-23 was also a consanguineous family with two RP affected females. These patients as well as three asymptomatic sibs were carriers for the C759F mutation whereas two additional unaffected sibs were C759F homozygotes. A thorough otological and ophthalmological investigation of these two subjects ruled out RP symptoms and hearing impairment, which may indicate that homozygosity for C759F does not cause disease.
USH2A has been shown to harbour mutations causing not only Usher syndrome type II,9,11–15 but also atypical Usher syndrome12,13 and non-syndromic ARRP.8 When the data reported here are considered, the phenotypic variation arising from different mutations in USH2A becomes extraordinarily pronounced, ranging from distinct types of Usher syndrome, through non-syndromic RP, to unaffected subjects.
There are many instances where different mutations in the same gene can result in diverse phenotypes. The USH2A gene is another example of such phenotypic variation of a Mendelian condition. In this work, we have described the presence of the missense C759F mutation associated with extreme phenotypic variation among unrelated subjects, that is, the symptomatic homozygous cases of family M-286 v the asymptomatic homozygous subjects from family S-23. A particularly striking example of phenotypic heterogeneity related to the USH2A gene is illustrated by the case of monozygotic twins who were homozygous for 2299delG.12
The possible mechanisms involved in this phenotypic variation are usually presented as assumptions or speculations and only exceptionally rely on proven data. One such exception has been the finding of triallelic inheritance in the Bardet-Biedl syndrome (BBS).16 BBS is a genetically heterogeneous disorder characterised by multiple clinical features, including pigmentary retinal dystrophy, which is considered to be an autosomal recessive condition. The authors reported the presence of three mutant alleles of the BB2 and BB6 genes in affected subjects. They also detected unaffected subjects who carried two BBS2 mutations but no BBS6 mutation, suggesting that BBS may not be a single gene recessive disease but a complex trait requiring three mutant alleles to manifest the phenotype. Based on this model, we can speculate that a similar phenomenon may underlie the extremely different phenotypes of the C759F homozygotes reported in the present work. These symptomatic homozygous patients would carry a third mutated allele of an unidentified gene whereas the asymptomatic homozygotes would lack the third abnormal allele required to manifest the disease. In line with the BBS model, C759F heterozygous symptomatic patients would carry two additional mutated alleles of the unidentified gene.
The wide range of phenotypes associated with usherin mutations underlines how the relationship between pathogenetic mutations and disease phenotype is becoming increasingly complex, rendering the Mendelian concept of monofactorial disease causation increasingly untenable for a growing number of diseases. Further molecular and biochemical studies to elucidate the function of this protein in the development and maintenance of the retina and cochlea will help to define the events that result in deafness and/or blindness and will provide insights into the physiology of vision and hearing.
Acknowledgments
This work was supported by grants from FIS (99-0010-03), ONCE/ Fundación ONCE, and Fundaluce. The authors belong to Grupo Multicéntrico Español para el Estudio de RP. We are grateful to Orland Diez for his helpful comments.