Article Text

Statistics from Altmetric.com
Editor—The hereditary spastic paraplegias (HSPs) are clinically characterised by progressive lower limb spasticity. The spasticity may occur in isolation (“pure”) or may be complicated by other major clinical features. Autosomal dominant, autosomal recessive, and X linked recessive inheritance patterns have been described for pure and complicated forms of HSP.1 ,2
There are loci for ADPHSP on chromosomes 2p (SPG4, MIM 182601),3 ,4 8q (SPG8, MIM 603563),5 ,6 14q (SPG3, MIM 182600),7 15q (SPG6, MIM 600363),8and 19q (SPG12).9 In addition, we recently mapped an ADPHSP locus on chromosome 12q13 (SPG10, MIM 604187) in a large UK family, family 4.10 This locus was narrowed to a 9.2 cM region between markers D12S368 and D12S83.
Clinical features and diagnostic criteria for family 4 have previously been described.10 ,11 Briefly, subjects were classified as being affected if they had lower limb hyperreflexia in addition to at least one of the following: progressive spastic gait abnormality, bilateral extensor plantar reflex, or bilateral sustained (⩾5 beats) ankle or knee clonus. Subjects were classified as being possibly affected if lower limb hyperreflexia was present without other abnormal signs and as being normal if they had an entirely normal neurological examination. Thirteen members of family 4 are affected by ADPHSP and the family has a relatively young mean age at onset of 10.8 (SD 9.6) years (range 8-40).
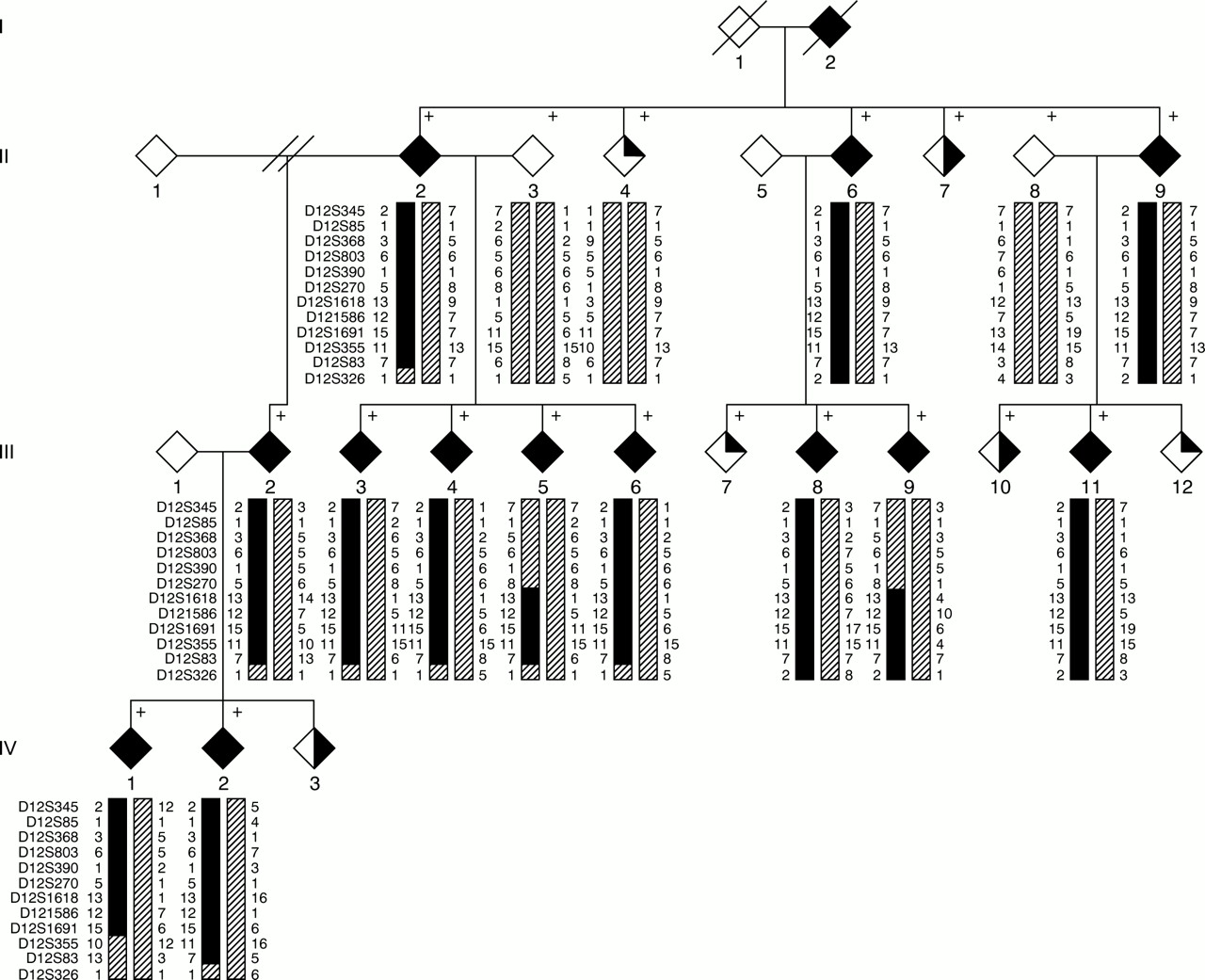
We have now genotyped additional markers D12S803, D12S390, D12S270, D12S1618, and D12S355 for subjects from family 4, using previously described methods.10 Primer sequences for these markers are available from the Généthon microsatellite linkage map12 or from the Marshfield Medical Research Foundation website. Haplotypes were constructed for these and for previously genotyped markers (fig 1). Obligate recombination events in two affected subjects (III.5 and III.9) redefine the centromeric boundary of the SPG10 region at D12S270, and an obligate recombinant event in affected subject IV.1 redefines the telomeric boundary at D12S355. These recombination events narrow the SPG10 candidate region to 6.95 cM (Marshfield Medical Research Foundation website).

Family tree of ADPHSP family 4, showing haplotypes for markers around the SPG10 region. The marker order is shown alongside each generation. For clarity non-contributory haplotypes are represented by a uniform hatched shading pattern. Recombination events in affected subjects narrow the SPG10 critical region to a 6.95 cM region between D12S270 and D12S335. Subjects' sexes have been anonymised for confidentiality reasons. Filled symbol=affected, half filled=possibly affected, quarter filled=clinically normal, +=DNA available.
A database search (Online Mendelian Inheritance in Man) was carried out to identify candidate genes within the SPG10 critical region. The neuronal sodium channel gene SCN8A is located at chromosome 12q13, close to marker D12S368.13 It is expressed in neurones throughout the central and peripheral nervous systems.14 ,15 Mice homozygous for null mutations inSCN8A (medand medtg ) develop progressive hind limb paralysis and muscle atrophy, with death in the juvenile period.14 ,16 The progressive paralysis in mouseSCN8A null mutants is caused by functional denervation of skeletal muscle, with normal motor neurone number and diameter.17 Although ADPHSP is pathologically characterised by “dying back” of the terminal ends of corticospinal tract axons,18 we considered there to be sufficient overlap between the ADPHSP phenotype and theSCN8A mouse null mutant phenotypes to investigate this gene as a candidate for the disease.
The 26 exons (with flanking intronic sequence) ofSCN8A were amplified by polymerase chain reaction, as previously described,13 using DNA from affected subject III.2. Each exon, with its flanking intronic sequence, was sequenced manually, as previously described,13 or with dye terminators (Perkin Elmer, Foster City) on an ABI377 automated sequencer (Perkin Elmer). Two previously identified single nucleotide polymorphisms were detected, a C to T in intron 19, position –27 from exon 20, and a C to T in exon 22, at position +90.13Neither of these variants change the amino acid sequence. We also detected a T for C substitution in exon 14 at position +175 (fig 2A). This new variant results in an arginine to cysteine substitution in the SCN8A protein at amino acid 1026, within a conserved region of the second cytoplasmic loop (fig 2B). The T for C substitution can be detected readily, since it abolishes anFnu4HI restriction site. We examined the segregation of this new variant in the family by carrying out PCR amplification of exon 14, digestion of PCR product with Fnu4HI, followed by agarose gel electrophoresis. The T for C variant segregated completely with the disease, except in affected family members III.5 and III.9, who were recombinants for this new SNP (fig 2C). These results excludeSCN8A from the SPG10 candidate region, with a likely placing centromeric to D12S1618. This location is consistent with the physical mapping of SCN8A to a YAC containing D12S368.13

{kind=link}
{kind=link}
Genetic variation in exon 14. (A) A 643 bp fragment containing exon 14 was amplified from genomic DNA with primers 14F (CAA GAG CCT CTT TGA GTC TGT CAC G) and 14R (ACA AGC ACC CTG TTT GCT CTC AC). Patient III.2 is heterozygous for a C to T transition within the exon (arrow), resulting in amino acid substitution R1026C and loss of an Fnu4HI restriction site. (B) R1026C is located in cytoplasmic loop 2 of the sodium channel SCN8A. Horizontal bars indicate intron/exon boundaries.13 (C) Wild type exon 14 contains Fnu4HI fragments of 116 bp and 134 bp which are replaced by a 250 bp fragment in the variant exon. Affected subjects III.6 and III.8 are heterozygous for the Fnu4HI site and generate all three fragments, but affected subjects III.5 and III.9 are homozygous for the wild type allele and do not generate the 250 bp fragment.
The frequency of the exon 14 SNP was determined by genotyping 80 parents from the CEPH pedigrees (CEPH website) and a panel of unrelated, white United Kingdom subjects, black African subjects, and Japanese subjects. The T for C substitution was present in 3/80 CEPH parents (3/160 chromosomes, 1.9%), 2/39 of the UK subjects (2/78 chromosomes, 2.6%), in 0/44 black African, and 0/27 Japanese subjects. To our knowledge, this is the first relatively common coding variant described within an ion channel gene. Further studies will be required to determine whether this polymorphism has any functional effect on the SCN8A protein, or whether it confers any predisposition to polygenic diseases which may arise from ion channel disorders.
To date, four different classes of genes have been implicated in HSP. X linked spastic paraplegia is caused by mutations in the cell adhesion molecule L1-CAM19 and mutations in a myelin gene, the proteolipid protein gene.20 One form of autosomal recessive HSP is caused by mutations in a nuclear encoded mitochondrial gene (paraplegin) which has metalloprotease and chaperone function.21 Finally, a gene for the most common form of ADPHSP, spastin, encodes a protein which may be involved in the structure of the nuclear proteosome.22 The diversity of HSP genes suggests that the neurodegeneration seen in HSPs may be a final common pathway for a number of processes, some of which may involve novel proteins. This makes selection of candidate genes problematical. Identification of additional SPG10 linked families will be crucial to narrow the candidate region and facilitate positional cloning of the SPG10 gene.
Acknowledgments
Electronic databases: Centre d'Etude du Polymorphisme Humain (CEPH) website: http://www.cephb.fr (for details of CEPH families). Marshfield Medical Research Foundation:http://www.marshmed.org/genetics (for marker genetic locations). Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/Omim(for HSP MIM entries and candidate gene search).
The first two authors contributed equally to this work. We thank the members of family 4 for taking part in this study. Marker genotyping and automated sequence analysis of SCN8A was carried out in the UK Medical Research Council HGMP Linkage Hotel. Patient assessment and sample collection was supported by the UK Medical Research Council. DCR is a Glaxo/Wellcome Research Fellow and ER is a Wellcome Research Training Fellow. ER is supported by a Sackler Fellowship.