Article Text

Statistics from Altmetric.com
The syndrome of persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) was described more than 40 years ago by Mc Quarrie.1 Despite the inordinate amount of interest in this syndrome, the pathogenesis of the disease has not yet been completely elucidated.
For decades, the disease has been ascribed to nesidioblastosis. This term, first coined by Laidlaw, describes the persistence of a diffuse and disseminated proliferation of islet cells budding off from ducts.2 The concept of nesidioblastosis as the underlying condition of hyperinsulinism is still deeply rooted in the mind of many clinicians, although it has been questioned by several authors.3-5 Indeed, observations based on quantitative immunohistochemical investigations have shown that nesidioblastosis is a common feature of the pancreas in normoglycaemic neonates and infants (fig 1).3

Nesidioblastosis in the pancreas of a normoglycaemic infant. B cells budding off from ducts stained by an anti-insulin antibody (immunoperoxidase; original magnification, ×140).
Progress in genetics and molecular biology has increased our understanding of the syndrome. By means of linkage analysis, hyperinsulinism in familial cases has been mapped to chromosome 11.6 Genetic molecular analyses have demonstrated mutations of the genes encoding both the sulphonylurea receptor7-10 and the Kir 6.2 subunit,11 as well as maternal loss of the imprinted gene at the 11p15 region, which might explain the insulin hypersecretion characteristic of this syndrome.12-14
In addition, physiological analysis has demonstrated the absence of functional ATP dependent potassium channels in some of these infants with hypoglycaemia.10 ,15
Several studies have clearly demonstrated the existence of two forms of PHHI. One corresponds to a focal pancreatic adenomatous hyperplasia (focal PHHI) and the other is characterised by a diffuse β cell abnormality (diffuse PHHI).16-18 Up until now, these two forms could not be differentiated by clinical or biochemical data, although their underlying pathological mechanisms and the treatment that they require are totally different. Indeed, the focal lesion is formed by the confluence of apparently normal islets and can be treated by resection limited to the focal lesion. In contrast, the diffuse form probably results from a generalised abnormal function of the β cells and its treatment requires a large or near total pancreatic resection.18
Some clinicians think that these two abnormalities are the expression of the same basic defect and continue to treat all patients with a near total pancreatectomy. However, in our opinion, such an extensive resection is not justified in cases with focal lesions.19
This review summarises our experience of PHHI based on the morphological analysis of the pancreas of about 100 neonates and infants with early onset hyperinsulinism. It is focused on the morphological heterogeneity of the disease and gives the keys for the differential diagnosis. The morphological characteristics of the focal form are reported and the pathogenesis of the diffuse form, as hypothesised from the morphological analysis, is discussed.
PHHI: not a single pathological entity
It is not possible to define a morphological feature that would be pathognomonic of the islets of infants suffering from PHHI. Indeed, the pathological examination of large series of such cases reveals that the syndrome of PHHI is not a single morphological entity. Certain cases are characterised by a focal lesion (fig 2), whereas others are devoid of any circumscribed lesion. This concept is now supported by other studies, such as in the field of molecular biology, and is of paramount importance from a clinical point of view.12-14 In our series, focal lesions represented about a third of the cases.

Focal lesion in a neonate. (A) Routinely stained section showing the focal lesion on the left part of the picture. The lobular structure of the pancreas is maintained (haematoxylin and eosin stained; magnification, ×15). (B) Immunohistochemistry with an anti-insulin antibody identifies more clearly the lesion that is formed by the confluence of apparently normal islets separated by few exocrine acini (immunoperoxidase; original magnification, ×15).
We have shown that the focal form of the disease can be cured by a partial resection restricted to the lesion, whereas a large resection or even a subtotal pancreatectomy is not always sufficient to cure patients with the diffuse form. The problem would be simple if it were possible to differentiate the two forms before surgery or to identify the focal lesion macroscopically during surgery. However, a specific preoperative differential diagnosis is often difficult and sometimes impossible.
No clinical or biological features are typical of infants affected by one or the other form of the disease. Furthermore, selective venous catheterisation with localised samplings for insulin measurements does not always allow the differentiation between the two forms. However, this method is the only one that can guide the surgeon and help to find and remove the pathological focus when hyperinsulinism is not related to a diffuse functional abnormality.20-22
Furthermore, in neonates and infants, it is usually difficult and most often impossible to identify macroscopically the focal lesion because of its small size and its structural characteristics. In fact, focal lesions in neonates are usually unlike the true adenomas seen in adults and children (that is, compact and easily detected by the surgeon) because they are formed by the confluence of apparently normal islets, which are sometimes separated by a few exocrine acini and maintain a lobular structure, as in the normal pancreas (fig 2).
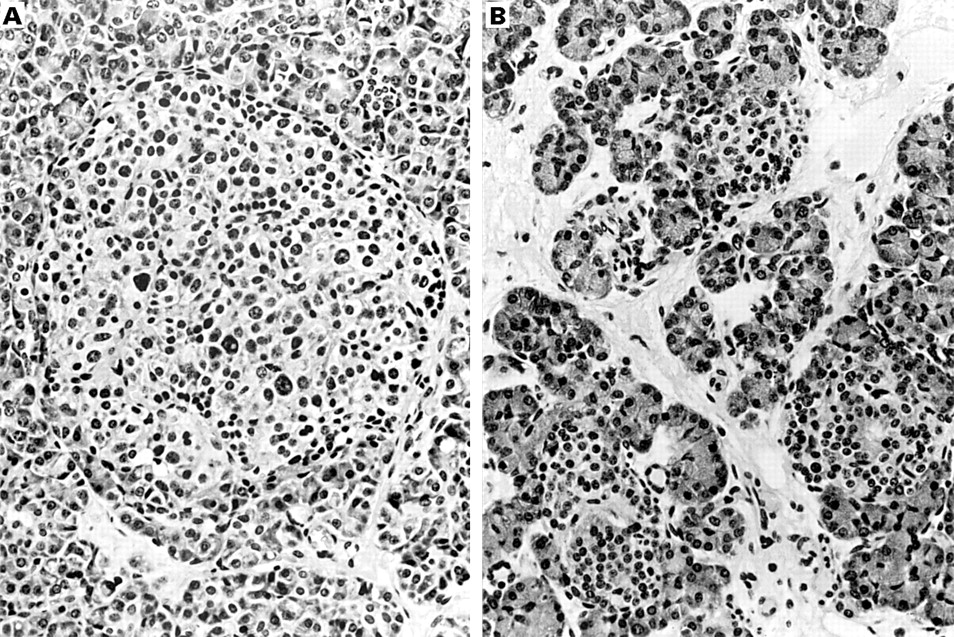
Histological analysis of diffuse and focal forms of the syndrome revealed that the diffuse form was associated with the presence of numerous abnormal large β cell nuclei (fig 3A) whereas, outside the lesion, no such nuclei were seen in the insular β cells of patients with a focal form of PHHI (fig 3B). Therefore, we analysed β cell nuclei and nucleo–cytoplasmic ratios in a large retrospective series to differentiate the two forms of the syndrome, in the hope that these criteria might be able to be used to determine whether hyperinsulinism was related to a focal or a diffuse form and to decide on the extent of pancreatectomy during surgery.
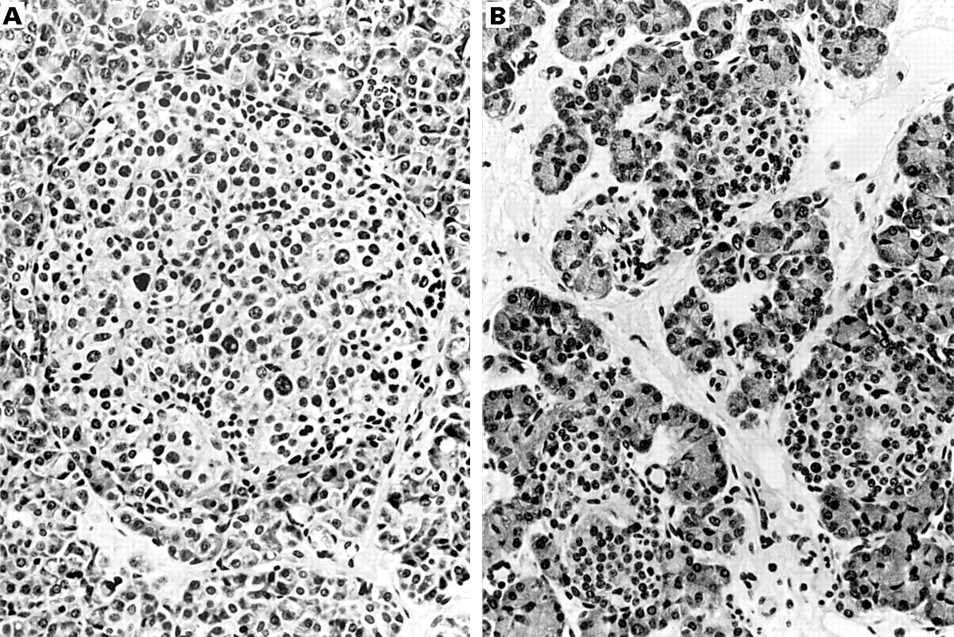
(A) Numerous abnormal large β cell nuclei in a pancreatic islet of an infant affected by the diffuse form of the syndrome (haematoxylin and eosin stained; magnification, ×420). (B) Normal β cell nuclei in the islets located outside the lesion in a focal form of the syndrome (haematoxylin and eosin stained; original magnification, ×420).
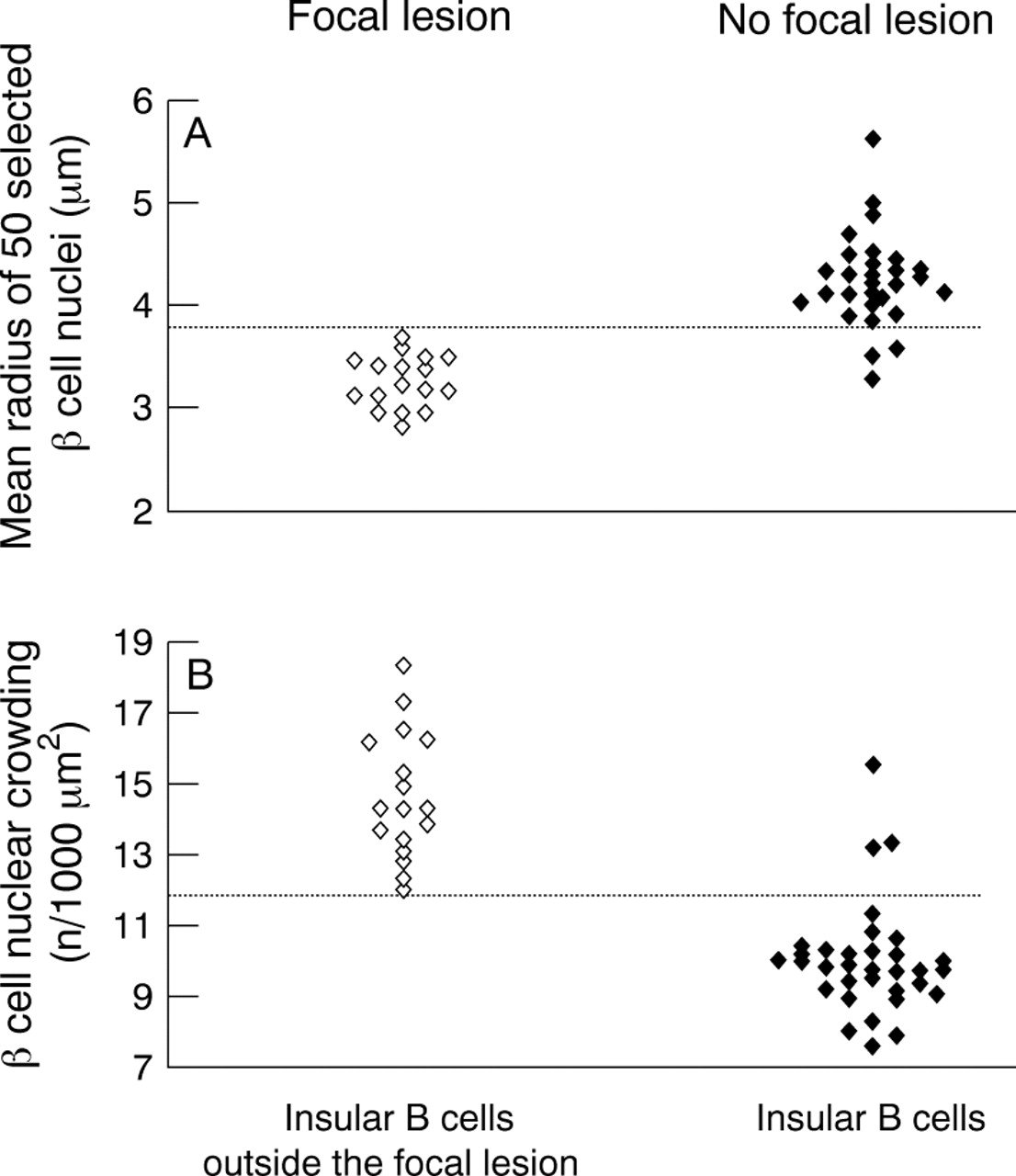
Conventional microscopy was used to search for the presence of abnormal β cell nuclei and to evaluate the nucleo–cytoplasmic ratio, and was combined with morphometry to analyse the characteristic differences between islets in both focal and diffuse forms of PHHI.17Two parameters were analysed: the size of the β cell nuclei (mean nuclear radius; MNR) measured with computerised image analyser, and the nucleo–cytoplasmic ratio of β cells evaluated by measuring the “β cell nuclear crowding” (BCNC, number of nuclei/1000 μm2 of β cell). Figure 4 clearly shows that both MNR and BCNC are significantly different in the two groups. Furthermore, these criteria are discriminant and allow the differentiation between the two groups because setting the threshold value of MNR at 3.7 μm and that of BCNC at 12.0 led to correct classification of 91% of the diffuse and 100% of the focal forms. Thus, β cell nuclear analysis could contribute to a subclassification of the syndrome, which cannot be based on clinical or biological data.
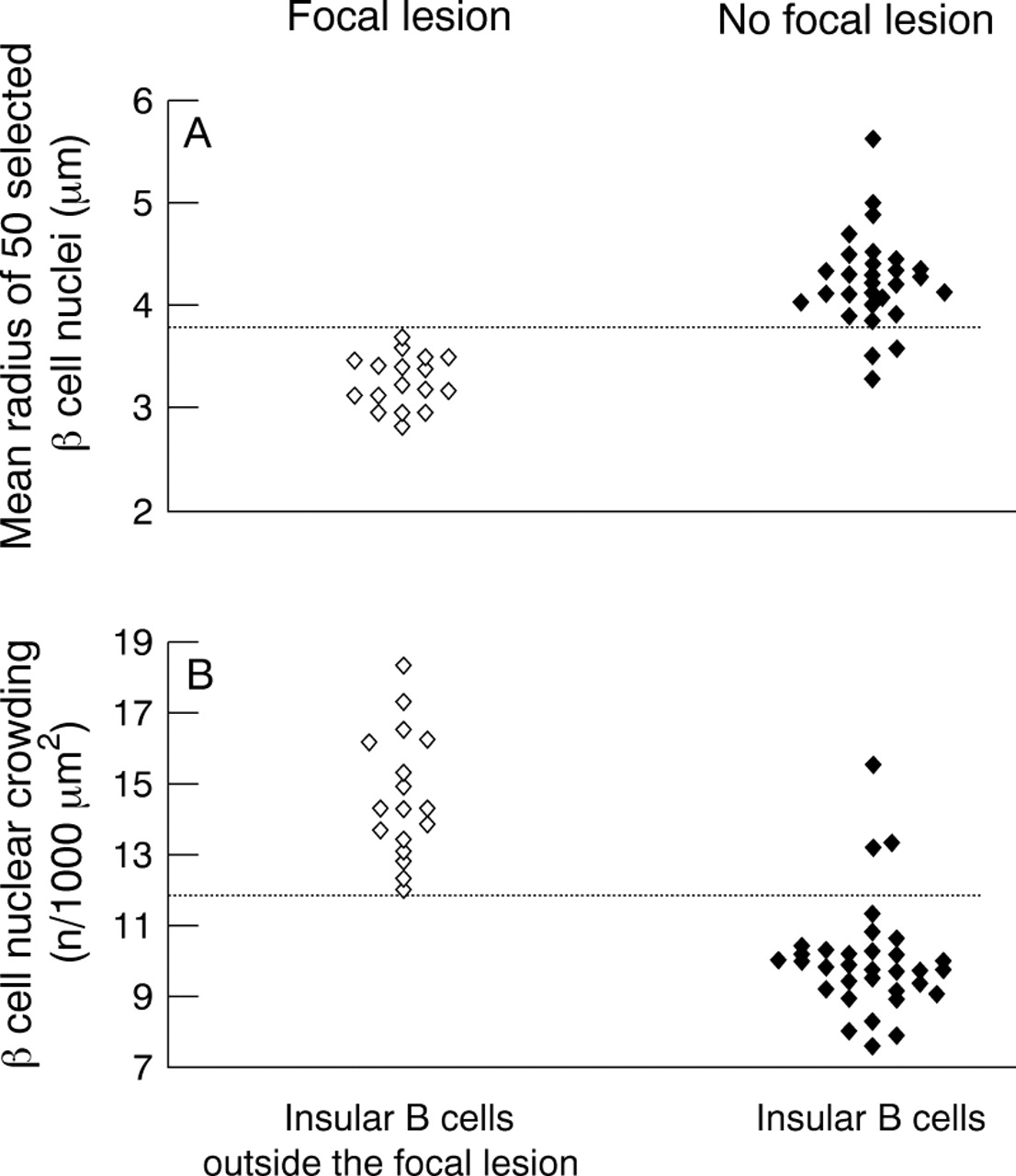
Mean nuclear radius (MNR; A) and β cell nuclear crowding (BCNC; B) enable the differentiation of the two groups at a threshold value (indicated by the broken line) of 3.7 μm for MNR and 12.0 for BCNC (modified from Sempoux and colleagues17).
In a second step, we investigated whether the distinction between normal and abnormal nuclei was possible during surgery.16The identification of abnormal β cell nuclei proved feasible, during surgery, on frozen sections of small pancreatic specimens taken from different parts of the gland. Thus, the morphological analysis of persurgical frozen sections might lead to the determination of the type of lesion (focal or diffuse), which would allow the surgeon to decide immediately during surgery on the most appropriate treatment (localised or extensive resection).
In summary, our studies demonstrated that two different forms of PHHI do exist. The diffuse form is associated with abnormal β cell nuclei and low nuclear crowding in the whole pancreas and requires a subtotal pancreatectomy. The focal form, characterised by the absence of abnormal β cell nuclei, can be cured by partial pancreatectomy restricted to the lesion. So far, only morphological examination of persurgical frozen sections has allowed us to distinguish between the two forms.
The focal form of PHHI: a β cell pancreatic tumour different from the “classic” insulinoma
The morphological and immunohistochemical analyses of a series of 27 focal lesions found in neonates and infants showed that this lesion was completely different from the insulinoma (also called adenoma) seen in children and adults. Insulinomas are usually macroscopically visible and are composed of β cells only arranged in nests or cords with a fibrovascular stroma. The cells and nuclei are often not polymorphous and mitoses are rare. In contrast, the focal lesion occurring in neonates is usually invisible to the naked eye, and is formed by the confluence of apparently normally organised islets, separated by a few exocrine acini, but maintaining a normal lobular pancreatic structure (fig 2).3 ,5 ,17 Endocrine cells are arranged in large clusters separated by a thin fibrovascular stroma or by small cords of acinar tissue. The nuclei appear polymorphous, and vary in size and chromatism. Mitoses may be numerous. Immunohistochemistry also identified A, D, and PP cells at the periphery of the endocrine clusters, as in normal islets, whereas β cells remain located in the centre.
The focal pancreatic lesion of infants is called focal adenomatous hyperplasia. In infants bearing such a lesion, hyperinsulinism results from the presence of the focal abnormality itself and, in the rest of the pancreas, the islets are often small but apparently retain a normal function because limited resection of the focal lesion restores normoglycaemia.18 Recently, de Lonlayet al have demonstrated a specific loss of the maternal allele located at 11p15, a chromosomal region submitted to parental imprinting, in focal adenomatous hyperplasia.12The somatic deletion of the maternal imprinted 11p15 region leads to a loss of H19, a tumour suppressor gene. In contrast, the IGF2 gene, which encodes insulin-like growth factor 2 (IGF-2), a growth factor suspected to play a role in pancreatic growth and tumorogenesis, is still expressed. Presumably, the abolition of H19 expression and the persistent expression of IGF2 could stimulate endocrine cell proliferation.
To test this hypothesis, we studied the proliferation rate of β cells in patients affected by PHHI and in age matched controls.23 A double immunohistochemical technique was used to detect Ki-67, a nuclear endogenous antigen present only during cell proliferation, and insulin as a pancreatic β cell marker. Our study clearly showed that in tumoral PHHI the labelling index (the percentage of Ki-67 labelled β cell nuclei) was significantly higher in cases of focal adenomatous hyperplasia than in either age matched controls or in adenomas, with the values of this last group not differing from those of age matched controls (fig 5). In contrast, the β cell labelling index was not significantly different in diffuse PHHI and in age matched controls.

Labelling index (number of Ki-67 labelled β cell nuclei/1000 β cell nuclei) of the different groups expressed as a percentage of age matched controls. In focal adenomatous hyperplasia, the labelling index is significantly higher than in controls and adenoma. In the diffuse form of the syndrome, the labelling index is not significantly different from that measured in age matched controls (modified from Sempoux and colleagues23). Adenomatous hyperplasia versus age matched controls, *p < 0.005; versus adenoma, **p < 0.025; versus diffuse PHHI, ***p < 0.01.
Thus, abnormal proliferation exists in adenomatous hyperplasia, which might be the result of an imbalance of H19/IGF2 gene expression. The demonstration of a normal proliferation rate of β cells in the diffuse form of the disease strongly argues against the hypothesis that nesidioblastosis is responsible for the hyperinsulinism in the non-focal form of the syndrome.
Pathogenesis of hyperinsulinism in infants without a focal lesion
The cause of hyperinsulinism in focal PHHI is easy to understand even though the mechanisms of insulin secretion by tumours are only partially known. Conversely, in infants with no circumscribed lesion, the cause of the hyperinsulinism is more difficult to determine because the pancreas appears to be normal, both macroscopically and microscopically.3 ,5
The first pathogenetic hypothesis evaluated was that of nesidioblastosis. The preliminary question was whether nesidioblastosis really exists. As shown in fig 1, nesidioblastosis defined as islets budding off from exocrine ducts does exist, but is also seen in strictly normoglycaemic control infants.3 ,5 In addition, the β cell proliferation rate is not higher in infants with diffuse PHHI than it is in control infants.23 Thus, nesidioblastosis exists, but is not specific for the syndrome of hyperinsulinism, and does not correspond to a continuous proliferation of endocrine cells.
The second hypothesis concerned a possible increase in β cell mass. It has been shown that the volume density of β cells evaluated by sensitive immunohistochemistry and reproducible morphometrical methods in large series of patients is not systematically higher in infants with hyperinsulinaemia than it is in controls (fig 6A).3Thus, neonatal hyperinsulinism is not related to increased β cell mass, which is hardly surprising because even large pancreatic resections sometimes fail to cure these patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Volume densities of β cells and D cells in diffuse PHHI and normoglycaemic controls. (A) β Cell volume density is not higher in PHHI. (B) D cell volume density is not significantly lower in PHHI than in controls (modified from Rahier and colleagues3).
The third hypothesis concerned a possible decrease in somatostatin producing cells (D cells).3 ,24 Because somatostatin inhibits β cells, a deficit in D cells could result in an excessive release of insulin. Because the volume density of somatostatin producing cells decreases rapidly with aging,16 it is crucial that control infants are strictly of the same age as infants with hypoglycaemia. The study of a large series of patients showed that the volume density of somatostatin cells tended to be lower in infants with hypoglycaemia than in controls (fig 6B).3 However, the differences were small and far from constant. Thus, it is unlikely that a global decrease in the volume density of somatostatin cells is responsible for the hypersecretion of insulin.
Because the hyperinsulinism could not be ascribed to nesidioblastosis, increased β cell mass, or decreased somatostatin cell mass, we hypothesised that it might result from abnormal β cell function.3 The demonstration of abnormal β cell nuclei in the pancreas of the infants affected by diffuse PHHI is in agreement with this hypothesis. Furthermore, the amount of proinsulin in the Golgi area is increased in the β cells of these infants, which is also indicative of a β cell hyperfunction.25 ,26
Finally, the demonstration of mutations in the genes encoding the sulfonylurea receptor7-10 and the potassium inward rectifier channel Kir 6.2,11 resulting in an abnormal function of ATP dependent potassium channels,10 ,15definitively confirms the hypothesis that, at least in certain patients, hyperinsulinism is primarily related to an abnormal β cell function.
From the heterogeneity of the morphological features of the islets, we are convinced that, even if one does not consider the problem of patients bearing a focal lesion, PHHI is not a single pathological entity. This is in agreement with certain recent reports.27 Further studies are in progress that hope to differentiate the various types of diffuse PHHI in close connection with a genetic analysis, with the hope that a better understanding of the pathogenesis of PHHI will lead to the improved management of these infants.
Acknowledgments
This study was supported by Grant 3.461593 from the Fonds de la Recherche Scientifique Médicale, Brussels, Belgium. The European Network for Research into Hyperinsulinism is supported by a concerted action grant (Biomed 2: grant number BMH4-CT98–3284). We thank Professor J-C Henquin for critical reading of the manuscript and V Delhaye-Liegeois for editorial assistance.