Article Text

Abstract
The understanding of hypertrophic cardiomyopathy (HCM) has changed dramatically over the last few decades, and it is now understood to be caused by a mutation in one of several cardiac sarcomeric genes. Due to complications such as outflow tract obstruction, diastolic dysfunction, arrhythmias, stroke, infective endocarditis and sudden cardiac death, appropriate and early identification of these patients is imperative. This review attempts to summarise the current state of knowledge on HCM, and provide insight of the appropriate investigations needed in patients with HCM. It also outlines treatment strategies for these patients. Much remains unknown about this complex and intriguing disease, and continued research in identifying the genetic basis of HCM, along with the assessment of therapeutic strategies, will help to optimise patient care.
Statistics from Altmetric.com
Cardiomyopathies can be classified into three major functional categories: dilated, restrictive and hypertrophic.1 Hypertrophic cardiomyopathy (HCM) may be distinguished from the other two types of cardiomyopathy by the presence of myocardial hypertrophy and, in approximately 25% of patients, left ventricular outflow tract obstruction (LVOTO).1 2 Originally described in 1869 as an asymmetric thickening of the septal myocardium,3 4 it was not until the 1950s that HCM emerged as a diagnostic entity.5–7 Currently, it is a complex heterogeneous disease caused by a genetic defect of the cardiac sarcomeric apparatus.7 8 Affecting 1 in 500 individuals in the general population,8–10 HCM is the most common inherited cardiovascular disorder.8 11–13 Complications, such as LVOTO, arrhythmias, diastolic dysfunction, myocardial infarction (MI), stroke, infective endocarditis (IE) and sudden cardiac death (SCD), make it essential to identify patients with this condition.8 14 The risk of death from HCM has been reported to be as high as 6% at tertiary care centres and at 1% in non-referred populations.2 8 14
PATHOLOGY
Gross
Hearts from patients with HCM commonly show cardiomegaly and left ventricular hypertrophy (LVH) (fig 1).8 Sixty-seven per cent of these hearts show asymmetric hypertrophy, involving predominantly the subaortic interventricular septum,7 8 but it may also be localised to other regions of the interventricular septum, and the free walls and apex of the left ventricle (LV).8 15 Isolated cases of right ventricular (RV) hypertrophy and biventricular involvement have also been reported.7 Symmetrical distribution of myocardial hypertrophy is seen in 10% of patients.1 HCM patients can have a wall thickness of between 15 and 30 mm (average 21–22 mm, normal <13 mm)8 16; the thickest wall ever reported was 60 mm.8

Saggital section of an explanted native heart demonstrating asymmetric myocardial thickening (arrows). The thickening is predominantly in the sub-aortic region. It may also occur in other areas of the heart, such as the mid-ventricular and apical regions, as well as the lateral and posterior wall. Right ventricular hypertrophy may be present in some patients. Apical aneurysms in the left (black dashed line) and right (white dashed line) ventricles are seen. The anterior mitral leaflet is large and thickened. LV, left ventricle; RV right ventricle.
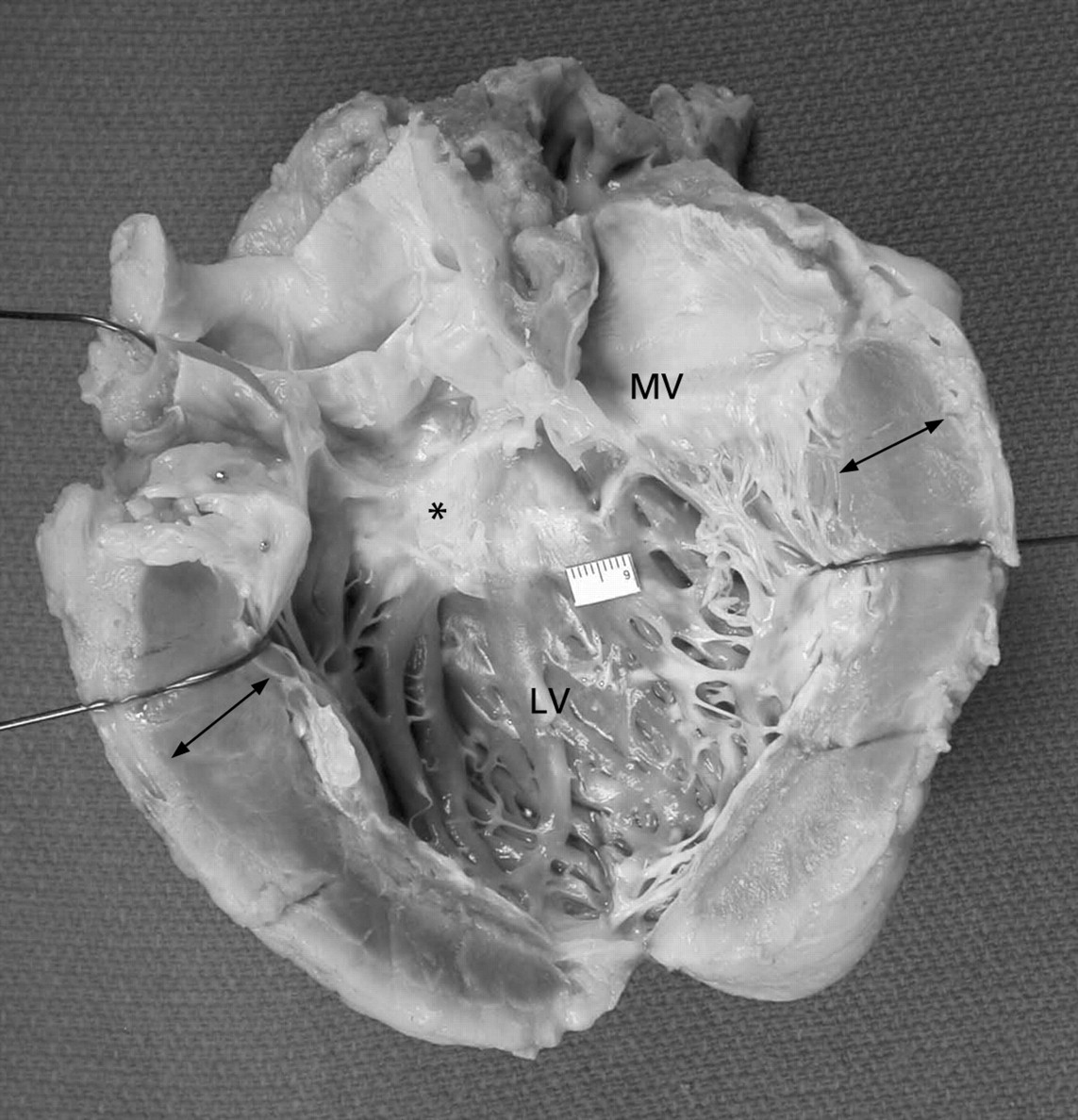
If the septum is involved, its bulging into the left ventricular cavity narrows the LV outflow tract and contributes to obstruction.15 17 LVOTO may be associated with endocardial and subendocardial fibrosis and thickening of the septum in the subaortic region due to repetitive contact (from hydrodynamic forces) between the septum and the anterior mitral leaflet during systole (fig 2).7 In fact, the mitral valve (MV) itself is abnormal (elongated and enlarged) in approximately two-thirds of patients.8 Anomalous insertion of the papillary muscles may infrequently be seen.8 18 19 These physical changes in the MV also contribute to the development of LVOTO, as the leaflets no longer coapt at their tips, but at the body of the leaflets during systole.
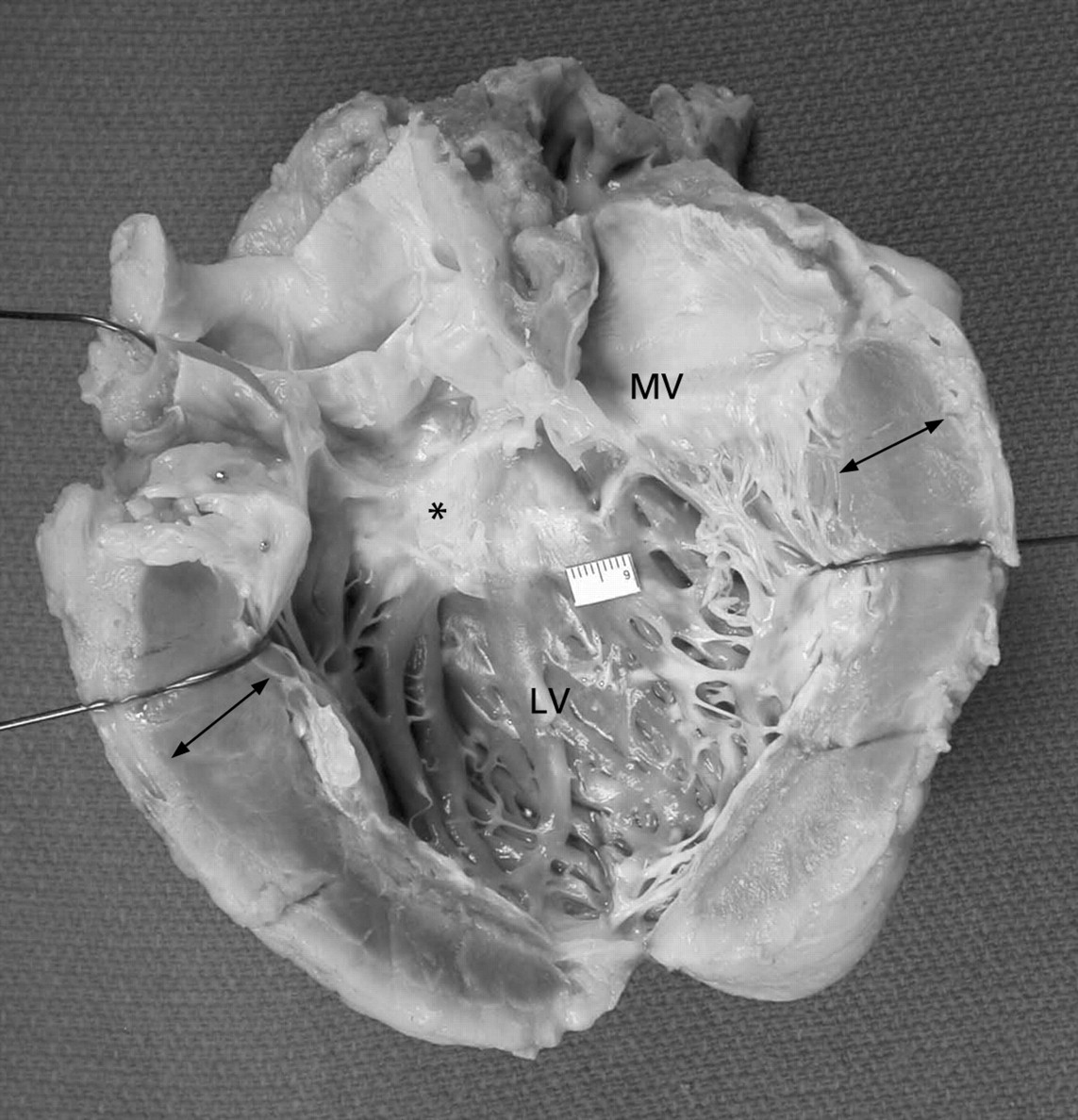
Explanted native heart showing the left ventricle (LV) and the LV inflow and outflow tracts opened. The ventricular walls are thickened (arrows). Subaortic septal fibrosis (*fibrotic systolic anterior motion (SAM) lesion) is seen. SAM lesions are formed by repetitive contact between the anterior mitral leaflet and the septum during systole. The mitral valve (MV) may also be abnormal (as in this case), with elongation, enlargement and fibrosis of the leaflets, and anomalous insertion of the papillary muscles. Together, the SAM lesion and the structural changes in the MV contribute to outflow obstruction.
Morphological changes in end-stage hypertrophic cardiomyopathy are characterised by dilatation of the ventricles and atria, and thinning of the walls due to myocardial scarring.20 21 Harris et al22 reported ventricular remodelling in 52% of patients with end-stage disease. A majority of these patients have end-diastolic LV cavities ⩾60 mm diameter (normal diameter <59 mm). Some patients may develop aneurysms in the apical region (fig 1). Similar changes may be seen on the RV and in the atria.22
Microscopic
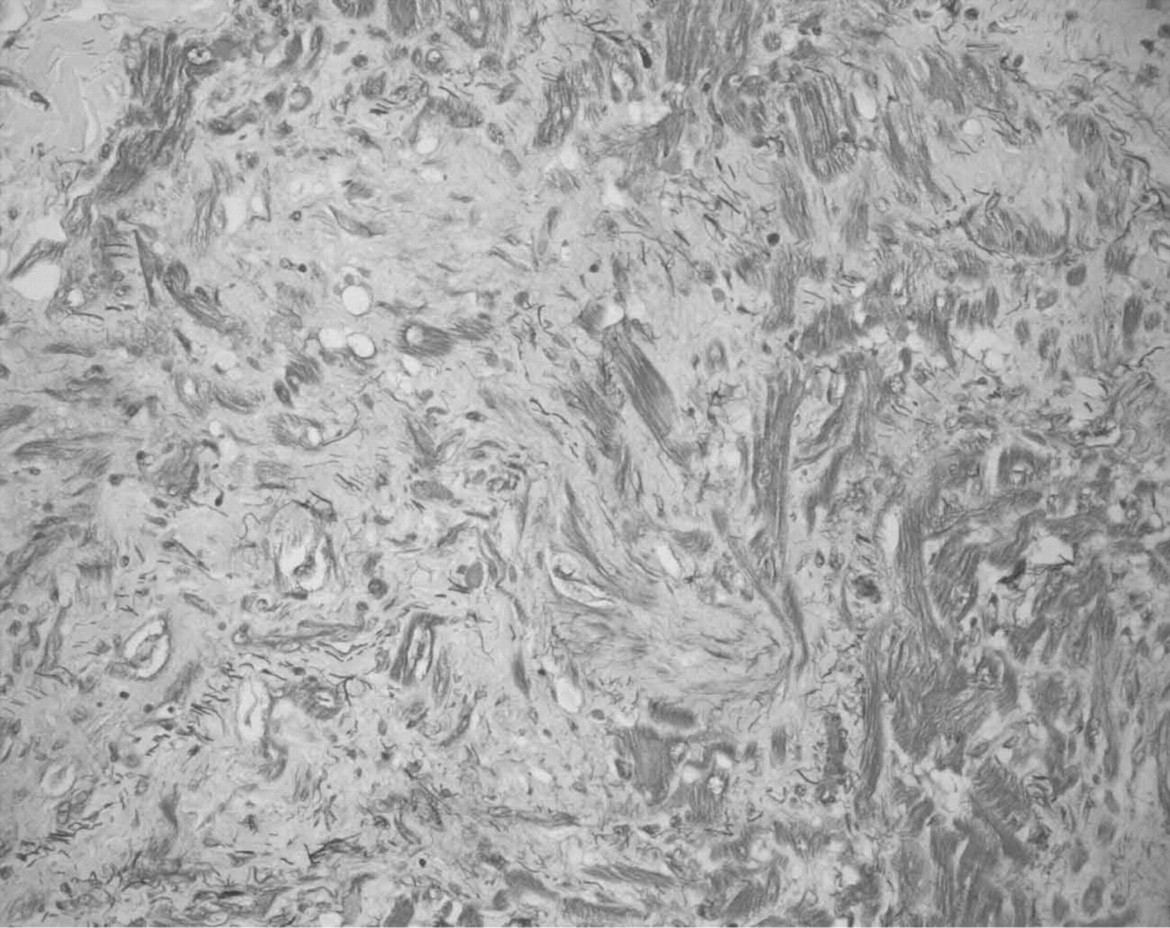
The most characteristic and common histological finding in HCM is myocyte disarray (fig 3A, B).1 8 Myocytes, normally arranged in straight parallel bundles, are found at oblique and perpendicular angles to each other, often in an interlacing or basket-weave pattern.7 8 Moreover, there are abnormal intercellular connections and variations in the diameter and length of individual myocytes (12–15 and 40 μm in diameter with normal and hypertrophied myocytes respectively).1 7 The hypertrophied myocytes are usually associated with interstitial myocardial fibrosis. The interstitium is characteristically loose and somewhat oedematous looking. As the disease progresses, the interstitium becomes collagen rich. There may also be variation in the size of myocyte nuclei.7 Similar histopathological changes may be observed in the atria.

(A) Microphotograph from the septal myocardium showing severe myocyte disarray. Normal myocytes run in straight, parallel bundles, but in hypertrophic cardiomyopathy, the myocytes are commonly found running at oblique angles. The individual myocytes vary in length, diameter and size of nuclei, and have abnormal interstitial connections. *Areas of significant interstitial myocardial fibrosis. Movat pentachrome stain, original magnification ×100. (B) Microphotograph of the myocardium showing severe myocyte disarray at high magnification. At this magnification, myofibril disarray (dashed line) is also evident in many of the myocytes. The myofibril disarray is usually a result of one of many possible sarcomeric gene mutations. At this magnification, variation in the size of nuclei is also more evident (Movat pentachrome stain, original magnification ×400).
Myocyte disarray is, however, not unique to HCM as it can occur in congenital heart disease, systemic hypertension and valvular aortic stenosis. Nonetheless, in comparison with these other disorders, myocyte disarray is far more extensive in HCM, involving as much as 40% of the entire myocardium (in order for the myocyte disarray to be considered significant, it should involve at least 20% of the myocardium in one histological section).8 23
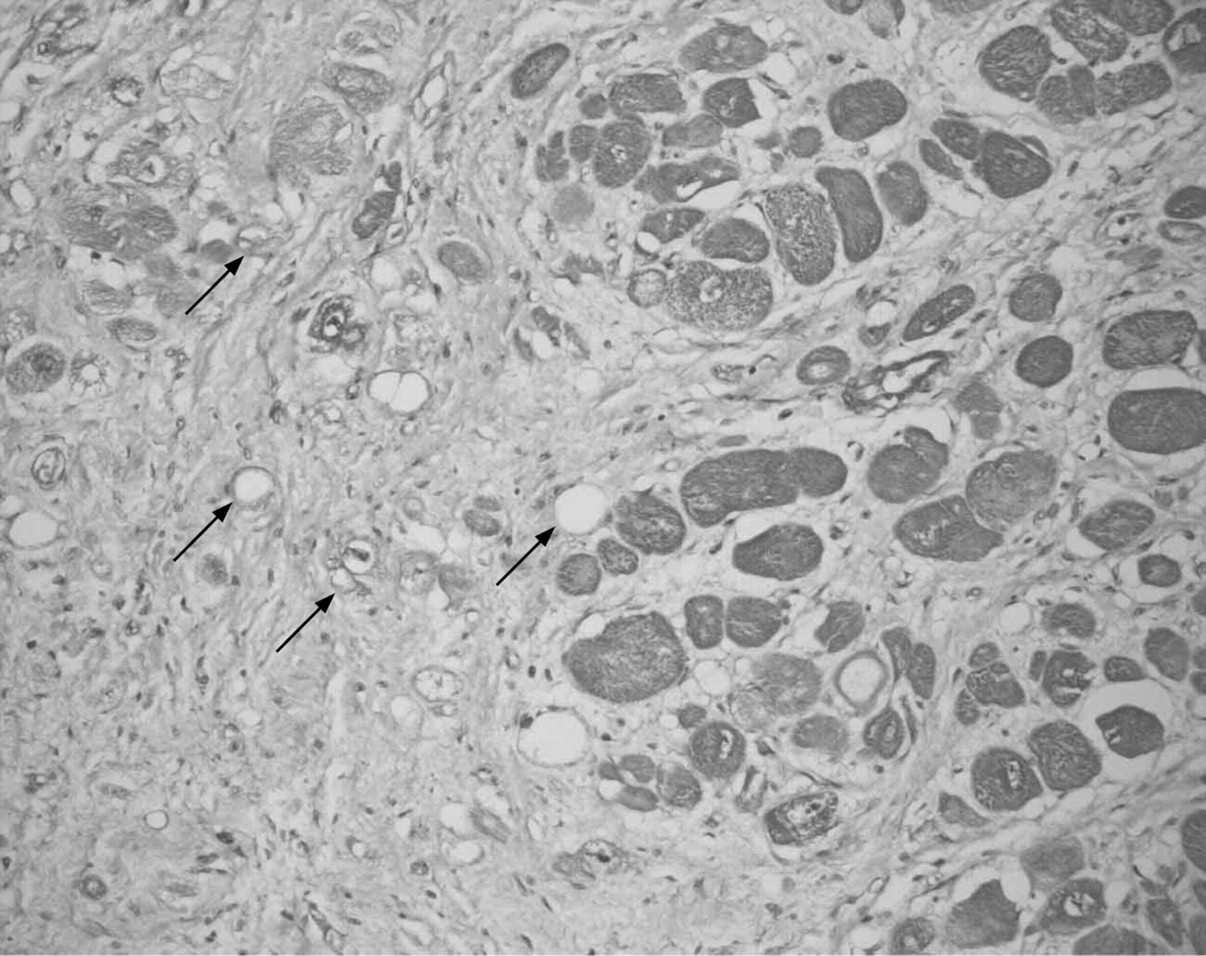
Abnormal intramural arteries are another common finding in the atria and ventricles (fig 4). These are characterised by medial thickening and narrowing of the lumen due to pressure from adjacent hypertrophied myocytes.24–26 This small vessel disease of intramural arteries is likely to contribute to ischaemia and resultant myocardial scarring,8 which predisposes certain individuals to ventricular arrhythmias.8 These vascular and myocardial changes are found in areas of visible hypertrophy, and they may also be found in areas that appear grossly normal. In patients with mutations in genes regulating myocyte metabolism, the typical features of myocyte disarray are often not seen. Instead these patients have non-membrane-bound vesicles containing glycogen and amylopectin.27

Microphotograph of an intramural artery with significant intimal thickening (arrows) with narrowing of the lumen (L). The lumen narrowing is increased by pressure from adjacent, hypertrophied myocytes. *Areas of medial fibrosis. Together, the intimal thickening, luminal narrowing and medial fibrosis lead to perfusion mismatches between myocardial demand and coronary supply, resulting in ischaemia and myocardial scarring. (Movat pentachrome stain, original magnification ×100).
End-stage disease is characterised by degeneration of muscle fibres and extensive fibrosis of the myocardium (⇓figs 5 and 6) and intramural arteries. Myocardial fibre disarray is also found to be more extensive and diffusely distributed (fig 7).9 20 21 These histopathological changes can be found in both sides of the heart, and in atria and ventricles. Evidence of extensive thinning of the myocardial walls may be found on histological analysis, characterised by a severe loss of myocytes (fig 8). Aneurysms may be identified histologically as well (fig 9).

Microphotograph of right ventricular wall of an end-stage hypertrophic cardiomyopathy. The walls are characterised by dilatation of the heart, and thinning of the walls due to multifocal myocardial scarring (*) and extensive interstitial fibrosis. Areas of fat infiltration (arrows) may also be seen (Movat pentachrome stain, original magnification ×16). E, epicardium.

Microphotograph of right ventricular wall of an end-stage hypertrophic cardiomyopathy. Many fibres show degenerative changes with large vacuoles and loss of myofibrils. The vacuoles (arrows) indicate dilatation of the sarcoplasmic reticulation and loss of normal microcellular structure. Large areas of fibrosis are seen (Movat pentachrome stain, original magnification ×200).
Microphotograph of right atrial myocardium of a hypertrophic cardiomyopathic heart with severe muscle fibre disarray and interstitial fibrosis. Muscle fibre disarray is the most diagnostic finding at histological analysis, and is responsible for weak contractile forces and impaired relaxation during diastole (Movat pentachrome stain, original magnification ×200).

Microphotograph of a dilated right atrial wall. There is marked loss of muscle fibres (arrows) in this section and replacement with connective tissue. This is part of the explanation for diastolic dysfunction and decreased contractility found in 80% of hypertrophic cardiomyopathy patients (Movat pentachrome stain, original magnification ×100). E, epicardium.

Microphotograph of left ventricular aneurysm (arrowheads) from a heart with cardiomyopathy. Aneurysms are most commonly found in end-stage hypertrophic cardiomyopathy (as opposed to early stage), especially in the apical regions due to high-pressure gradients compared with other areas of the heart. Transmural fibrosis, fat infiltration and interstitial fibrosis are seen in adjacent areas. Myocyte disarray is also noted (Movat pentachrome, original magnification ×16). E, epicardium.
BIOCHEMICAL CHANGES
Calcium
Abnormal calcium homeostasis is observed during active contraction in patients with HCM. These abnormalities are associated with impaired or asynchronous LV relaxation. This affects early diastolic ventricular filling and may be associated with delayed after-depolarisation.7
Natriuretic peptides
Atrial natriuretic peptide (ANP) is normally released from the atria in response to increased atrial volume. Brain natriuretic peptide (BNP) is released from the ventricles in response to increased ventricular pressure and/or volume. In HCM, plasma levels of ANP can be up to five times their normal level, while plasma levels of BNP can be up to 85 times normal.7 BNP levels are associated with symptomatic status in this condition and there is a wide variability in the levels of BNP detected in patients with HCM.28 HCM is also characterised by an abnormal secretion of ANP from the ventricles.7
Fatty acid metabolism
Abnormal fatty acid (FA) metabolism has been reported in several HCM studies. These studies have shown abnormal myocardial FA uptake and reduced expression of CD36, which is associated with long-chain FA transport across cellular membranes.7
AETIOLOGY: GENETICS
Several genes have been implicated in HCM and account for 60–70% of all cases of HCM (table 1).7 These genes encode proteins of the cardiac sarcomeric apparatus and exhibit an autosomal dominant pattern of inheritance.8 27 Patients with mutations in two additional non-sarcomeric genes may present with increased wall thickness mimicking HCM.29–31
Box 1: Genetically determined disorders that may present with hypertrophic cardiomyopathy
Syndrome
Friedreich ataxia
Lentiginosis
Noonan syndrome
Sywer syndrome
Metabolic disorder
Acyl-coenzyme A dehydrogenase deficiency
Carnitine deficiency
Debrancher enzyme deficiency
Fabry disease
Glycogen storage disease
Pompe disease
Forbes disease
Hurler syndrome
Hunter syndrome
Mitochondrial cytopathies
Sarcomeric genes
The most commonly mutated sarcomeric genes causing HCM are the β-myosin heavy chain (MYH7) and the cardiac myosin-binding protein C (MYBPC3) genes, which account for over 50% of all cases of HCM.8 The genetic diversity is compounded by intragenic heterogeneity, with over 400 different mutations identified within these 11 genes.8 27 The majority of mutations are missense or minor truncations with single amino acid substitutions.8 The majority of mutations occur in the myosin head or head–rod junction of the protein, suggesting that they may alter critical functional domains of the β-myosin heavy chain proteins that are involved in contractility.32 In contrast, mutations of the MYBPC3 gene have been associated with late disease onset and a better prognosis. In a study by Niimura et al,33 only 58% of patients under the age of 50 years with a mutation involving the MYBPC3 gene had evidence of HCM, and penetrance was incomplete even at 60 years of age. Numerous studies of genotype–phenotype correlations in HCM over the past 15 years have been inconclusive and genetic testing is currently not routinely used in the risk stratification of patients with HCM.7 11 34 35
Non-sarcomeric genes mimicking HCM
Recently, two genes responsible for myocardial metabolism have been identified as mimicking HCM. These genes include lysosome-associated membrane protein-2-α-galactosidase (LAMP2) and adenosine-monophosphate-activated protein kinase (PRKGA2).29–31 These mutations alter myocardial metabolism, resulting in increased wall thickness, cardiac storage abnormalities and conduction irregularities.29 Patients with these mutations are classified as patients with storage disorders.29
Other genetic disorders that cause hypertrophy
There are other genetic disorders that may present with increased left ventricular wall thickness (box 1). Most of these present during infancy and childhood, although some such as Fabry and mitochondrial cytopathies may manifest in adulthood.7
CLINICAL PRESENTATION
The most common presenting symptoms in patients with HCM include dyspnoea, angina, and presyncope or syncope. Chest pain may be precipitated by exertion or following a heavy meal (in patients with LVOTO) or it may occur at rest. Syncope may be caused by a number of mechanisms, including atrial or ventricular arrhythmias or significant LVOTO.7 Patients with obstructive HCM may be identified on physical examination.36 The jugular venous waveform may demonstrate a prominent “a” wave. The carotid upstroke may be brisk or double (traditionally called the “spike and dome” carotid upstroke). Palpation of the precordium may reveal a palpable fourth heart sound, a sustained apical impulse, or a triple apical impulse (the latter only seen in patients with LVOTO). Auscultatory findings include reversed splitting of the second heart sound and a dynamic systolic ejection murmur. In addition to manoeuvres that accentuate the murmur, include assuming the standing position (after squatting) and performing the Valsalva manoeuvre. There may also be an associated systolic murmur of mitral regurgitation.
INVESTIGATIONS
The diagnosis of HCM is most frequently made at echocardiography (table 2).2 Most commonly, HCM must be distinguished from hypertensive heart disease and infiltrative processes such as amyloidosis.1
Electrocardiogram
Approximately 75–90% of HCM patients have an abnormal ECG.8 Some of the abnormalities seen are a result of LVH and include voltage abnormalities, ST segment and T wave abnormalities, prominent Q waves, and signs of left atrial (LA) enlargement.8 37–40 The ECG finding of giant negative T waves in the precordial leads is characteristic of apical HCM and was first described in Japan.7 41 Arrhythmias may also be detected.
Two-dimensional echocardiography
Two-dimensional echocardiography (TTE) remains the most readily accessible and informative tool in making the diagnosis of HCM.2 8 42 Some of the classic TTE findings of HCM include asymmetric septal hypertrophy, a small LV cavity, and LA enlargement. TEE findings suggestive of obstruction include systolic anterior motion (SAM) of the anterior (or posterior) mitral leaflets and mid-systolic notching of the aortic valve.43 Echo-Doppler assessment is also useful in the assessment of diastolic function.
Doppler assessment
Doppler recordings, done at rest or during activity, measure LVOT pressure gradients and quantify jets of mitral regurgitation.8 The LVOT gradient is dynamic and may be assessed at rest and/or following provocative manoeuvres such as performing the Valsalva and inhaling amyl nitrate. Mitral regurgitation in patients with HCM characteristically consists of a posteriorly directed jet caused by SAM of the anterior mitral leaflet and the resultant interleaflet gap between the anterior and posterior mitral leaflets.44
Magnetic resonance imaging
MRI is useful when hypertrophy is confined to unusual areas, such as the apex, which may be difficult to image adequately with echocardiography in some patients. Contrast agents such as gadolinium may be used with MRI to identify regions of interstitial fibrosis and/or scarring.45 46
Cardiac catheterisation
Cardiac catheterisation is useful to exclude epicardial coronary artery disease as a cause of angina in patients with risk factors for coronary atherosclerosis.7 It can also be used to measure LV outflow tract gradients (at rest and with provocation) in patients with LVOTO.
Radionuclide studies
Radionuclide studies are potentially helpful in identifying areas with perfusion abnormalities. They can be used to identify areas of ischaemia.7
Cardiopulmonary exercise testing
Exercise testing is a useful means for assessing patients with HCM. Characteristic findings include a reduced peak oxygen uptake and anaerobic threshold.47 It can also be used to distinguish between respiratory and circulatory causes of exercise intolerance. Testing may reveal impaired cardiac output, or increased ventilatory carbon dioxide due to increased physiological dead space, abnormal microvascular function or abnormal respiratory stimulation.7 However, these findings are not specific to HCM, as increased ventilatory carbon dioxide may also be seen in non-HCM patients with congestive heart failure. Respiratory gas analysis upon exertion can also be used to assess the extent of diastolic dysfunction and disease severity. In a study comparing 135 HCM patients with 50 healthy individuals, oxygen consumption upon exertion was less than predicted in HCM patients compared with controls, and this finding was more pronounced in patients with obstructive disease.48
Endomyocardial biopsy
Patients with suspected HCM may uncommonly require an endomyocardial biopsy (EMB), which is useful in differentiating between different causes of increased LV wall thickness.49 For example, an EMB may be useful to differentiate between HCM, infiltrative disorders such as amyloidosis, and storage disorders such as Fabry disease, and patient treatment can therefore be adjusted accordingly.7 The EMB can help establish the diagnosis of HCM, since muscle fibre hypertrophy and muscle fibre disarray are very often present in the immediate subendocardium and not just in the deeper parts of the LV wall (septum or free wall) (J Butany, personal communication, 2008).
Genetic testing and family screening
There are still many difficulties in translating gene-based research into practical clinical strategies that could be used on a routine and widely available basis. It is difficult to identify the specific genetic defect in a given individual, since there are over 400 mutations associated with HCM. Genetic testing can potentially detect the specific mutation in 50–60% of patients2 42 Once a mutation is confirmed in the proband, the identification of this mutation in other family members is possible. Since routine genetic testing is not available and HCM exhibits an autosomal-dominant pattern of inheritance, it is recommended that first-degree relatives under the age of 20 be screened regularly (every 2 years prior to adolescence and more frequently during adolescence). After the age of 25 years, these individuals should be screened every 5 years.42
CLINICAL IMPLICATIONS
Athlete’s heart
It may be difficult to differentiate between normal myocardial adaptations to athletic activity and the morphological changes associated with HCM.7 8 50 51 The diagnosis of HCM is more likely than the diagnosis of athlete’s heart in the presence of the following: (1) a family history of HCM, (2) bizarre ECG patterns, (3) asymmetric hypertrophy (or other unusual pattern of left ventricular hypertrophy), (4) a small left ventricular cavity size (<45 mm), (5) LA enlargement, and (6) an abnormal left ventricular filling pattern.51 Finally, hypertrophy secondary to athleticism is more likely in individuals participating in endurance sports.
Hypertrophic cardiomyopathy in the elderly
HCM in the elderly has a more favourable prognosis and a lower risk of SCD compared with younger patients with HCM.52–55 While both groups exhibit myocardial hypertrophy, the LV cavity of younger patients tends to be more crescent shaped, compared with a more ovoid cavity in elderly patients.7 56 Another characteristic feature of HCM in the elderly is a decreased angle between the aorta and the long axis of the LV. This, along with age-related shrinkage of the heart, is believed to result in an upper-septal bulge (known as a “sigmoid septum”) and consequent obstruction of the outflow tract.7
PATHOPHYSIOLOGY
Diastolic dysfunction
Diastolic dysfunction is found in the majority of patients with HCM.8 57 Impaired diastolic function is associated with increased filling pressures and a non-compliant LV.8 57 58 LV relaxation is altered by myocyte disarray, interstitial fibrosis, hypertrophy, abnormal intracellular calcium flux and myocardial ischaemia.7 Impairment in LV relaxation and/or decreased ventricular compliance ultimately results in atrial enlargement and increased arial pressure, which manifest as dyspnoea and exercise intolerance.7
LV outflow tract obstruction
LVOTO is an independent determinant of heart failure and HCM-related death.8 14 LVOTO is a dynamic process, sometimes manifesting only during active movement or after pharmacological interventions.8 Twenty-five per cent of individuals with HCM exhibit an outflow gradient at rest alone, and this gradient may vary with volume status, alcohol intake, or consumption of a heavy meal.8 14 36
Arrhythmias
Atrial fibrillation (AF) occurs in 25% of all patients with HCM, and tends to increase in frequency with age.8 59 AF is a major determinant of embolic stroke and other peripheral vascular events and can also cause acute clinical deterioration due to reduced diastolic filling and cardiac output.8 Ventricular arrhythmias, although not as common as AF, are of great clinical importance as they are the most common cause of SCD.7
Myocardial ischaemia
Myocardial ischaemia reflects the interaction of multiple pathophysiological mechanisms, including reduced arteriolar density, myocardial fibrosis, myocyte disarray and diastolic dysfunction.8 A reduction in arterial density relative to the extent of LVH results in a mismatch between myocardial oxygen demand and supply. There is an increase in oxygen demand by the hypertrophied myocytes themselves, but a decrease in supply due to diastolic dysfunction. As a result, a decrease in coronary perfusion has been linked to syncope, abnormal blood pressure response during exercise, predisposition to ischaemia, LV systolic and diastolic dysfunction, and SCD.7 8 Small-vessel disease contributes to myocardial ischaemia, and its severity is a clinical indicator of disease progression in patients with HCM.7 60 A recent study by Cecchi et al60 demonstrated that the degree of microvascular dysfunction in HCM patients is an independent predictor of heart failure symptoms and death.
Sudden cardiac death
The most common cause of SCD is ventricular tachyarrhythmias. Risk factors for SCD include a previous history of cardiac arrest, a malignant family history of HCM with SCD, episodes of recurrent unexplained syncope, an abnormal blood pressure response with exercise (in young adults), non-sustained ventricular tachycardia, and a maximum LV wall thickness of at least 30 mm.42 The incidence of SCD is felt to be in the range of 1% in adult patients with HCM42 although it has been reported to be as high as 3% in HCM patients with multiple risk factors.61 62
TREATMENT
Counselling
Prior to any treatment strategy, patients should be counselled on family screening and activity levels (fig 10). Since HCM follows an autosomal dominant pattern of inheritance, informing patients of screening protocols for first-degree relatives may readily identify asymptomatic affected individuals. Further, patients should be counselled to avoid the extremes of exertion.64

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment algorithm for patients newly diagnosed with hypertrophic cardiomyopathy. Treatment varies depending on symptoms and severity of disease. AICD automatic implantable cardioverter defibrillator; AV, atrioventricular; IE, infective endocarditis; LVOT(O), left ventricular outflow tract (obstruction); SCD, sudden cardiac death.
Left ventricular outflow tract obstruction
There are four treatment strategies for the management of LVOTO in HCM: pharmacotherapy, surgical myectomy, septal ethanol ablation (SEA) and dual-chamber permanent pacing (DDD) (table 3).7
Medical treatment should be attempted in patients presenting with obstructive symptoms such as severe angina, dyspnoea, and/or syncope.2 8 The first line of treatment involves the use of negative inotropic agents (eg, beta blockers and/or disopyramide).2 8 65–67 Verapamil may potentially also be used in this condition but it has significant vasodilatory properties and should be used with extreme caution in patients with obstructive HCM (and with appropriate monitoring).2 7 8 17 29 68 69 ACE inhibitors should be avoided in HCM patients, as they may provoke or accentuate LVOTO.8
Patients who do not respond to medical treatment are referred for an invasive intervention. Surgical myectomy was the first therapy developed for this condition and it is still considered as a treatment modality.2 8 70 Myectomy is performed via a transaortic approach and approximately 5–15 g subaortic myocardium is removed from the LV outflow tract. Operative complications are rare, but include complete heart block (requiring a permanent pacemaker), ventricular septal defect, aortic regurgitation, and left bundle branch block.8 71 Operative mortality is less than 1% for isolated myectomy.2 8 71 72
Over the last decade, SEA has emerged as a viable treatment option for the management of patients with drug-refractory obstructive HCM.8 65 Approximately 1–3 ml absolute ethanol is injected into the first septal perforator branch of the left anterior descending artery. This forms a localised area of myocardial necrosis, with resulting thinning of the septal wall, enlargement of the LVOT, and elimination of SAM and of outflow obstruction.7 8 73–78 Complications such as ablation of inappropriate areas of the myocardium can be minimised by peri-interventional use of myocardial contrast echocardiography.7 42 It may take approximately 6–12 months before symptomatic improvements are seen.7 79 Because ablation has only recently been introduced, extended follow-up studies have not been performed.65 80
DDD pacing is an option for relieving symptoms of LVOTO without removing the physical obstruction itself. However, data on its clinical efficacy are contradictory, and it is usually reserved for patients who are not candidates for either surgical myectomy or SEA.2 42
Arrhythmias
Patients with HCM usually become symptomatic with the onset of atrial fibrillation.2 29 68 Physicians should also attempt to control heart rate using calcium channel and beta blockers.2 Amiodarone may be effective in the long-term management of atrial fibrillation.7 81
Sudden cardiac death
Patients with sustained ventricular arrhythmias are at increased risk of SCD.7 82 83 Recent studies have demonstrated the efficacy of implantable cardioverter defibrillators (ICDs) in the primary and secondary prevention of SCD.2 84 These devices have been shown to be highly effective in preventing arrhythmias, and their benefits are superior to amiodarone.7 8 84 However, ICDs are associated with a high incidence of inappropriate ICD discharges.85
Symptomatic and non-symptomatic non-obstructive HCM
There are few data available on the treatment of symptomatic non-obstructive HCM. Calcium channel and beta blockers may be effective in relieving dyspnoea and angina, as well as increasing exercise tolerance.8 While nitrates are relatively contraindicated in patients with LVOTO, they can be effective in patients with angina and breathlessness in the absence of obstruction. Diuretic treatment may be helpful in relieving symptoms of congestive heart failure.7
Patients with non-symptomatic, non-obstructive HCM should not be given any treatment. Instead, disease progression should be monitored using ECG and echocardiography, as outlined above.
CONCLUSIONS
The understanding of HCM has changed dramatically over the last two decades. HCM is now recognised as a genetically inherited, autosomal dominant, sarcomeric protein-associated disease. Its genetic heterogeneity is matched by its variable clinical and morphological presentation, and although universal screening is not yet feasible, it is imperative that physicians perform appropriate tests to accurately identify patients (and asymptomatic relatives) with suspected HCM. Much still remains to be clarified about this complex disease. Continuing research in identifying de novo disease-causing mutations, along with the testing of novel therapeutic modalities, will help optimise patient care and improve prognosis, hopefully ensuring an adequate quality of life in these patients. More analyses and publication of studies on morphological tissues (hearts and myectomy specimens) of patients with HCM are essential to provide greater correlations between the morphological changes (especially interstitial fibrosis and mural vascular changes) and the clinical manifestations of this condition.
Acknowledgments
The authors would like to thanks Melissa Skarban, Shaun Leong and Lisa Hurlock for their assistance in completing this manuscript.
REFERENCES
Footnotes
Competing interests: None.