Article Text

Abstract
Background In chicken, loss of TALPID3 results in non-functional cilia and short-rib polydactyly syndrome. This phenotype is caused by a frameshift mutation in the chicken ortholog of the human KIAA0586 gene, which encodes a novel coiled-coil domain protein essential for primary ciliogenesis, suggesting that KIAA0586 can be associated with ciliopathy in human beings.
Methods In our patients with ciliopathy (http://www.clinicaltrials.gov: NCT00068224), we have collected extensive clinical and neuroimaging data from affected individuals, and performed whole exome sequencing on DNA from affected individuals and their parents. We analysed gene expression on fibroblast cell line, and determined the effect of gene mutation on ciliogenesis in cells derived from patients.
Results We identified biallelic mutations in the human TALPID3 ortholog, KIAA0586, in six children with findings of overlapping Jeune and Joubert syndromes. Fibroblasts cultured from one of the patients with Jeune–Joubert syndrome exhibited more severe cilia defects than fibroblasts from patients with only Joubert syndrome; this difference was reflected in KIAA0586 RNA expression levels. Rescue of the cilia defect with full-length wild type KIAA0586 indicated a causal link between cilia formation and KIAA0586 function.
Conclusions Our results show that biallelic deleterious mutations in KIAA0586 lead to Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Furthermore, our results confirm that KIAA0586/TALPID3 is essential in cilia formation in human beings, expand the KIAA0586 phenotype to include features of Jeune syndrome and provide a pathogenetic connection between Joubert and Jeune syndromes, based on aberrant ciliogenesis.
- ciliopathy
- Clinical genetics
- Developmental
- whole exome sequencing
- small thorax
Statistics from Altmetric.com
Introduction
The chicken TALPID3 mutation, originally identified in 1964 on the basis of its eponymous limb defects,1 disrupts transduction of sonic hedgehog activity in the limbs, the neural tube and the somites.2 ,3 This phenotype is caused by a frameshift mutation in the chicken ortholog of the human KIAA0586 gene, which encodes a novel coiled-coil domain protein essential for primary ciliogenesis.2 ,4 Hedgehog signalling is a highly conserved pathway that plays a fundamental role in animal development.5
Joubert syndrome is a ciliopathy characterised by distinctive midbrain and cerebellar malformations that result in the ‘molar tooth sign’ on axial brain images as well as hypotonia and developmental delay.6 The majority of patients with Joubert syndrome also display episodic tachypnoea or apnoea and abnormal eye movements. The term ‘Joubert syndrome and related disorders’ (JSRD) is used to describe individuals with Joubert syndrome plus other findings, including retinal dystrophy, ocular colobomas, hepatic fibrosis and fibrocystic renal disease. JSRD are a genetically heterogeneous group of ciliopathies; the 25 causative genes identified to date account for approximately 50%–70% of cases.
Jeune syndrome, also known as asphyxiating thoracic dystrophy, is a skeletal ciliopathy characterised by a small thorax due to short ribs, disproportionate short stature due to short long bones, polydactyly and renal, hepatic and retinal involvement.7 Radiological skeletal abnormalities include cone-shaped epiphyses in the hands and feet, irregular metaphyses, decreased iliac cephalocaudal height and a trident-shaped acetabulum.8 To date, biallelic mutations in 12 genes, including CEP120, IFT80, DYNC2H1, TTC21B and WDR19, have been found to cause Jeune syndrome.
Many ciliopathies, including Joubert, Bardet–Biedl and Meckel–Gruber syndromes, display genotypic and phenotypic overlap; this has not been demonstrated for patients with Joubert syndrome with KIAA0586 mutations, however. Here we present six patients with biallelic mutations in KIAA0586 and brain anomalies classic for Joubert syndrome, two of whom manifest a Joubert–Jeune overlap ciliopathy with the additional finding of a small thorax and respiratory problems.
Materials and methods
Patients
Patients and parents were evaluated at the National Institutes of Health (NIH) Clinical Center and enrolled in the NIH protocol ‘Clinical and Molecular Investigations into Ciliopathies’ (http://www.clinicaltrials.gov, trial NCT00068224), approved by the Institutional Review Board of the National Human Genome Research Institute (NHGRI). The parents gave written, informed consent. Evaluations at the NIH Clinical Center included family history and physical examination and comprehensive biochemical and imaging studies.
Imaging
All patients underwent axial, coronal and sagittal T1-weighted or T2-weighted brain MRI imaging at their local hospitals as part of their routine diagnostic evaluation. All available brain MRI studies were retrospectively evaluated for infratentorial and supratentorial abnormalities as previously reported.7 Cerebellar dysplasia was defined as an abnormal cerebellar foliation, fissuration and white matter arborisation. Standard and high-resolution ultrasonographic studies were performed using 4 and 7 Mhz transducers (AVI Sequoia, Mountain View, California, USA).
The images were reviewed by two paediatric neuroradiologists, an adult neuroradiologist, a geneticist and a paediatric neurologist with high expertise in cerebellar malformations.
Sequencing and variant analysis
For exome sequencing, we used the HiSeq2000 (Illumina)8 that employed 101 bp paired-end read sequencing. Image analysis and base calling were performed using Illumina Genome Analyzer Pipeline software V.1.13.48.0 with default parameters. Reads were aligned to a human reference sequence (UCSC assembly hg19, NCBI build 37) using a package called Efficient Large-scale Alignment of Nucleotide Databases (Illumina, San Diego, California, USA). Genotypes were called at all positions where there were high-quality sequence bases using a Bayesian algorithm called the Most Probable Genotype,9 and variants were filtered using the graphical software tool VarSifter V.1.5.10 The database dbSNP (http://www.ncbi.nlm.nih.gov/snp/) covers the 1.22% of the human genome corresponding to the Consensus Conserved Domain Sequences and more than 1000 non-coding RNAs.11 BAM files of the subjects were visualised using Integrative Genomics Viewer (Broad Institute).
For dideoxy sequencing, primers were designed to cover all coding exons and flanking intronic regions of KIAA0586 (primer sequences available on request). Direct sequencing of the PCR amplification products was carried out using BigDye V.3.1 Terminator chemistry (Applied Biosystems) and separated on an ABI 3130xl genetic analyser (Applied Biosystems). Data were evaluated using Sequencher V.5.0 software (Gene Codes, Ann Arbor, Michigan, USA).
Cell culture
Fibroblasts were cultured from forearm skin biopsies and grown in high-glucose (4.5 g/L) DMEM supplemented with 15% FBS, 2 mM L-glutamine, non-essential amino acid solution and penicillin-streptomycin. For control, normal adult human dermal fibroblasts (ATCC PCS-201-012) were used.
Expression studies
Total RNA was isolated from fibroblasts with RNeasy Mini kit (Qiagen, Valencia, California, USA)12 and from whole blood using PAXgene Blood RNA Kit. RNA was treated with a DNase kit (DNA-free) according to the manufacturer's protocol (Applied Biosystems, Austin, Texas, USA). RNA concentration and purity were assessed on a Qubit Fluorometer (Life Technologies). First strand cDNA was synthesised using a high-capacity RNA-to-cDNA kit (Applied Biosystems). Human multiple tissue cDNA panels (human MTC panels I and II, and Human Fetal MT Panel) were purchased from Clontech (Clontech Laboratories, Mountain View, California, USA). Quantitative real-time PCR (qPCR) was performed using a KIAA0586 (Hs00914952_m1(Probe 1) and Hs0091496_m1 (Probe 2)), Assays-On-Demand primer-probe assays (Applied Biosystems) and a control assay for the ACTB gene. PCR amplifications were performed on 100 ng of cDNA using TaqMan Gene Expression Master Mix reagent (Applied Biosystems), and were carried out on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems). Isoform analysis was made using primers as detailed in online supplementary table S1. qPCR was performed using a Bio-Rad iQ SYBR Green Supermix and qPCR machine with standard qPCR parameters to analyse the expression of KIAA0586 isoforms compared with the control gene POLR2A. Results were analysed with the comparative CT method as described.13 ,14
RT-PCR and TA cloning
RNA from patient 1 and his parents were isolated, and cDNA synthesis was performed as mentioned above. RT reaction products were diluted and amplified using specific primers (primer sequences available on request) as shown in online supplementary figure S1A (upper panel). PCR products were loaded into 2% agarose gel and visualised on ultraviolet transilluminator. In order to confirm the deletion, PCR products (from P1, P1-f and P1-m) were cloned in TOPO vector according to manufacturer's instructions. Random colonies were picked up, and the desired region was sequenced.
Immunofluorescence microscopy
Antibodies that were used in this study were: rabbit anti-ARL13B antibody (Proteintech Group, Chicago, Illinois, USA), mouse anti-gamma tubulin (clone GTU-88, Sigma), Alexa Fluor secondary donkey antirabbit and donkey antimouse antibodies (Invitrogen).
For cilia analysis, cells were grown in coverslips until 70% confluent, serum-starved for 24 h and fixed using ice-cold methanol for 10 min. After three washes in PBS, cells were blocked with 2% donkey serum and 2% BSA in PBS, and incubated in primary antibodies overnight at 4°C. After washing, samples were incubated in appropriate secondary antibodies, washed and mounted in Vectashield containing DAPI (Vector Laboratories). Cells were imaged with a Zeiss 510 META confocal laser-scanning microscope. Optical sections were collected from the xy plane and merged into maximum projection images. A total of 200 cells were analysed per cell line. Non-parametric t test was used to compare control and patient cells.
Production of lentiviral vectors and transduction of cells
cDNA encoding the full-length human KIAA0586 (NM_001244189.1) was obtained by RT-PCR-mediated cloning, using total RNA from testes. KIAA0586 was cloned into pENTR11 vector by Seamless Cloning, and later subcloned into pLenti6.3/V5-DEST (Invitrogen) using Gateway Recombination (Invitrogen). Production of lentiviral constructs was performed by transient cotransfection of 293FT cells (ViraPower Lentiviral Expression Systems, Invitrogen), producing pLenti6.3-KIAA0586 and pLenti6.3-empty for control, and concentrated using Lenti-X Concentrator (Clontech) following manufacturer's protocol to generate high titre virus. Fibroblasts cells (control, patient 1 and 2) were transduced with viral preparations (1×107/mL, at an MOI of 10) diluted in fresh culture media supplemented with Polybrene (Sigma, with a final concentration of 8 µg/mL) twice over the duration of 12 h. Forty-eight hours after transduction, generation of cells stably expressing pLenti6.3-KIAA0586 was initiated by changing the culture media with selection antibiotic (Blasticidin, Invitrogen). After serial dilution and clone selection for stable cell line generation, cells were used for assays.
Results
KIAA0586 mutations in patients with Jeune and Joubert syndromes
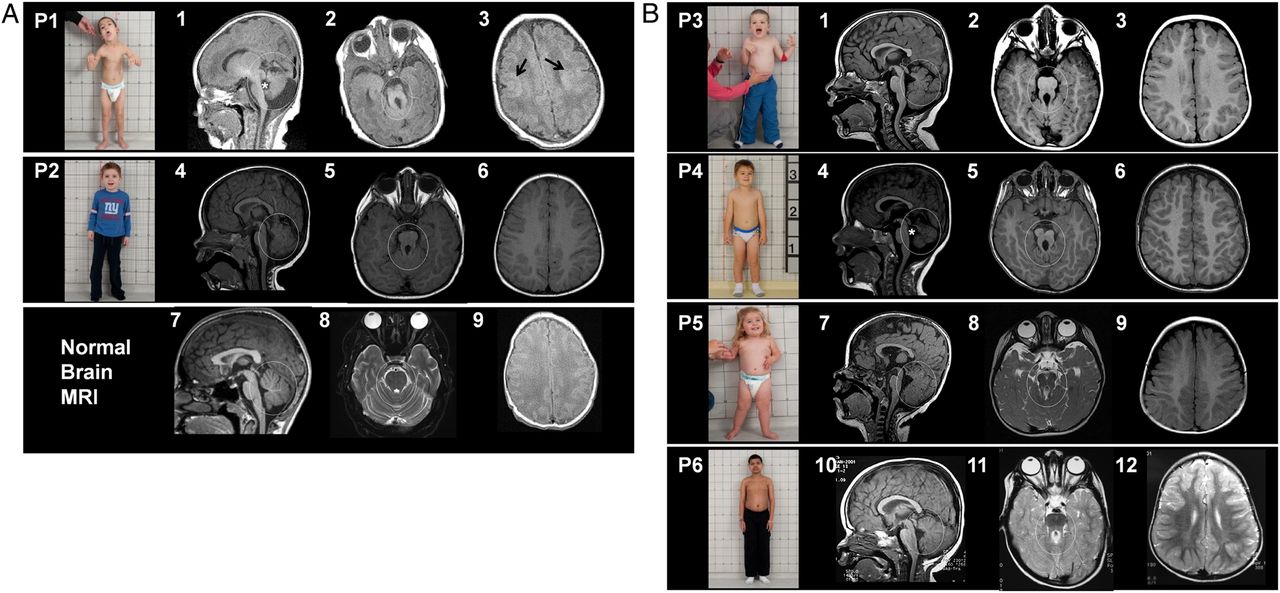
Patient 1 (figure 1A, upper panel, P1; table 1) was evaluated at the NIH Clinical Center at age 4.7 and 6.8 years. Prenatal ultrasonography showed Dandy–Walker anomaly at 22 weeks’ gestation. At birth, patient 1 had respiratory distress associated with a small bell-shaped thorax; he spent 12 weeks in a neonatal intensive care unit due to respiratory distress, central and obstructive apnoea and intermittent tachypnoea. Brain MRI was most consistent with Dandy–Walker variant with cerebellar and midbrain anomalies similar to Joubert syndrome (figure 1A1,A2), and bilateral diffuse polymicrogyria of supratentorial grey matter (figure 1A3). The infant was discharged with the diagnoses of Joubert and Jeune syndromes. He required several readmissions due to respiratory distress and hypoventilation. He had poor feeding due to oral motor dyscoordination and episodic tachypnoea, and required 100% G-tube feeding till age 6.5 years. At 6.8 years, his chest size had increased, but still remained small (circumference ∼54 cm, 10th percentile). There was global developmental delay; he walked at 5.5 years, and was non-verbal at age 6.7 years. He had disproportionate short stature with relatively short extremities (figure 1A; upper panel, P1; table 1). He had bilateral fifth finger clinodactyly and overlapping toes; there was no polydactyly or syndactyly (see online supplementary figure S1A). Eye examination revealed oculomotor apraxia and limited lateral gaze bilaterally, and retinal examination was normal. Optic nerve head pallor indicating optic atrophy was noted on examination (see online supplementary figure S1B).
Clinical characteristics of patients with KIAA0586 mutations

Clinical photographs and imaging of subjects with Jeune–Joubert syndrome and Joubert syndrome. (A) Upper panel shows photograph of patient 1 (P1) with small thorax, relatively long trunk and short extremities, and craniofacial dysmorphism, including low set incompletely rotated ears, hypoplastic midface, epicanthal folds and lower lip scars due to biting. Brain MRI performed on the third day of life showing severe cerebellar vermis hypoplasia and dysplasia, marked retrocerebellar fluid (circle) and enlarged fourth ventricle with abnormal shape (asterisk) in (1); thickened superior cerebellar peduncles with abnormally oblique orientation resulting in the ‘molar tooth sign’(circle) in (2) and cerebral cortex showing bilateral diffuse polymicrogyria (arrows) in (3). Middle panel shows photograph of patient 2 (P2) with normalised chest size at age 4.4 years; his chest was small and bell-shaped during the first year of life. Brain MRI at 18 months showing a mildly hypoplastic and dysplastic vermis (circle) in (4) and molar tooth sign (circle) in (5) and normal cerebral cortex (6). Lower panels show normal brain MRI images for comparison (7, 8 and 9). (B) First panel shows photograph of patient 3 (P3) displaying epicanthal folds, low set ears and normal chest size. Brain MRI at 2 years 4 months displaying a hypoplastic and dysplastic vermis (circle) in (1), molar tooth sign (circle) in (2) and normal cerebellar cortex (3). Second panel shows photograph of patient 4 (P4) showing normal chest size. Brain MRI of P4 at age 3 years 3 months displaying severely hypoplastic and dysplastic vermis (circle) and enlarged and abnormally shaped fourth ventricle (asterisk) in (4), molar tooth sign (5) and normal cerebral cortex (6). Third panel shows photograph of patient 5 (P5) showing a chubby 26-month-old with normal chest size. Brain MRI of P5 at age 14 months showing mildly hypoplastic and dysplastic cerebellar vermis (circle) and enlarged fourth ventricle (7), molar tooth sign (circle) in (8) and normal cerebral cortex (9). Fourth panel shows patient 6 (P6), whose chest size was normal. Brain MRI of P6 shows mildly hypoplastic and dysplastic cerebellar vermis (10, circle), enlarged fourth ventricle images, molar tooth sign (circle, in 11) and normal cerebral cortex (12).
Patient 2 was evaluated at age 4.4 years (figure 1A, middle panel, P2; table 1). Perinatal history was uneventful. At 6 months, he required hospitalisation due to respiratory distress during a viral respiratory infection. He was noted to have a small bell-shaped chest, hypotonia and developmental delay. MRI at 18 months showed a mildly hypoplastic and dysplastic vermis (marked in circle in figure 1A4) and a molar tooth sign (figure 1A5). By 4.4 years of age, his chest size had normalised (figure 1A, middle panel, P2) and his height was within the normal range (table 1). Unlike patient 1, patient 2 had a normal cerebral cortex (figure 1A6). Eye examination showed a unilateral form fruste coloboma of the retina and subtle optic nerve head pallor bilaterally. Liver and kidney-related blood and urine chemistries were normal in both patients. Abdominal ultrasonography was normal in both patients except for mildly elevated liver echogenicity.
Exome sequencing was performed on both patients 1 and 2 and their parents. We identified two variants in KIAA0586 (NM_001244189.1) and confirmed them using Sanger sequencing. Patient 1 had a synonymous variant inherited from his father (KIAA0586: c.990C>T; p.Leu330Leu), and patient 2 had a frameshifting duplication inherited from his father (KIAA0586: c.130dupC; p.His44Profs*8) (figure 2A). The synonymous p.Leu330Leu (exon 9) resulted in aberrant splicing leading to the loss of exon 9 (see online supplementary figure S2), as detected in RNA derived from patient-1 fibroblasts (see online supplementary figure S2A). By cloning the bands detected by amplifying regions between exon 8 and exon 10, we show that this deletion removes 154 bp at the RNA level (see online supplementary figure S2B), producing a frameshift with early termination 3 amino acids after the deletion in exon 10, and causing partial or complete loss of function. The c.130dupC (p.His44Profs*8) from patient 2 is predicted to cause an early termination. An additional analysis of the BAM files (see online supplementary figure S3) revealed a potential deletion in both families affecting multiple exons. We then designed multiple primer pairs to identify the breakpoints of this deletion by PCR and Sanger sequencing (figure 2A). This deletion, c.745-350_1288+1117del8260 bp, affects exon 8, 9 and 10, removes 544 coding bp and leads to a frameshift deletion with an early termination 3 amino acids after the deletion in exon 11. Analysis of this deletion in 190 samples from ethnically matched unaffected controls showed that there were no alleles that were either homozygous or heterozygous (data not shown).
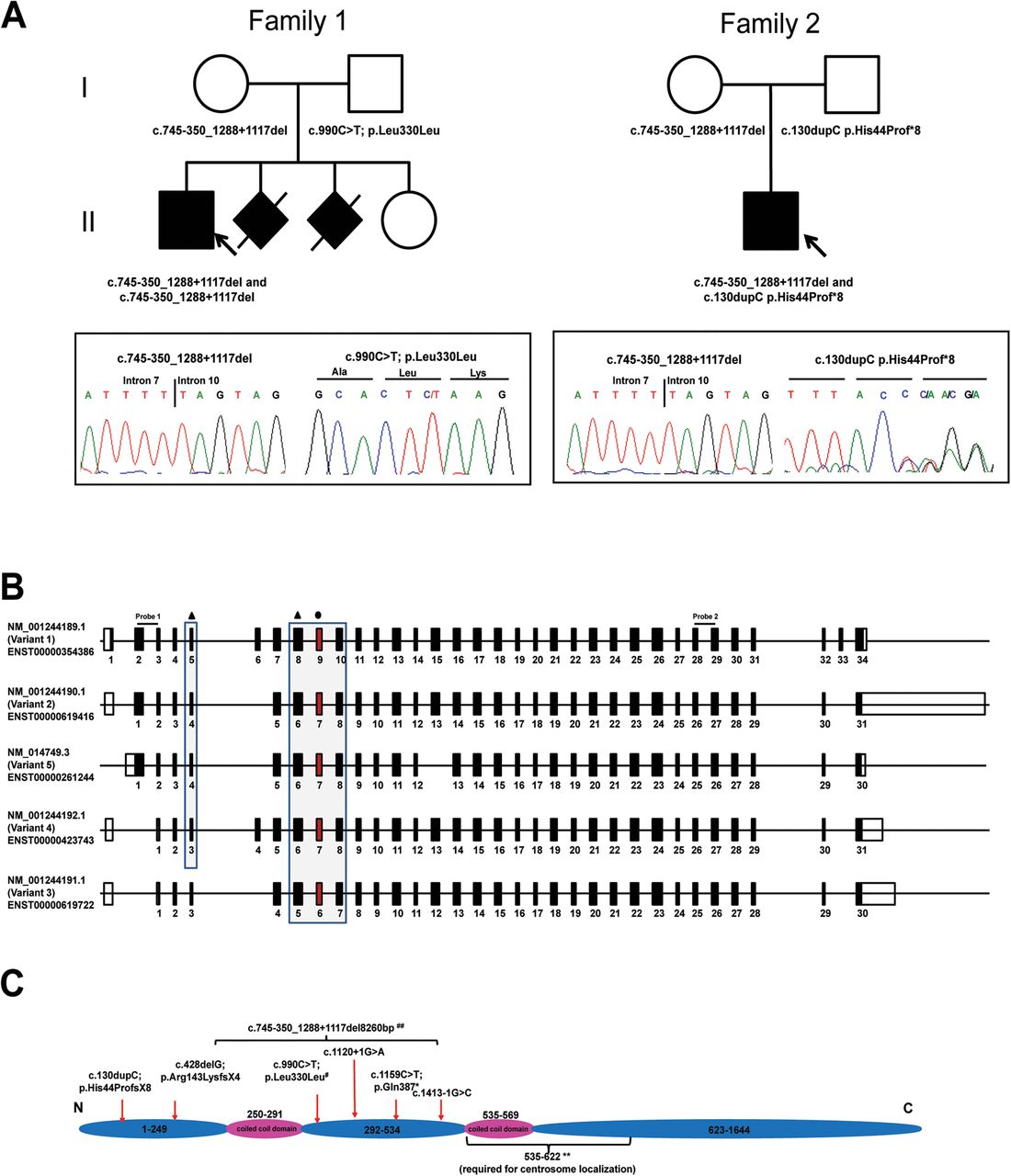
Pedigree of families and molecular data. (A) Family pedigrees of patient 1 and patient 2 and representative chromatograms showing variants in KIAA0586. Affected individuals are shown in black, while arrows point to the probands in each family. Note that in patient 1, the c.990C>T mutation appears homozygous; patient 1 is hemizygous for this variant, as the other allele on this region is deleted due to the mutation that he inherited from his mother (c.745-350_1288+1117del). (B) Schema showing the main isoforms of KIAA0586. Mutations affect most, but not all, isoforms. Black triangle refers to mutations that are predicted to produce a premature stop; black circle refers to the synonymous variant that leads to deletion of exon 9 (exon highlighted in red). Probe 1 and probe 2 refers the exons amplified for expression analysis. (C) Protein schema showing the location of mutations. All mutations are found before amino acids 535-622, a highly conserved domain required for centrosome localisation (**, refers to the findings by Yin et al4). # refers to the synonymous variant that leads to the deletion of exon 9; ## refers to the variant that deletes exon 8, 9 and 10.
We then performed exome analysis on 130 samples of our multisystem ciliopathy families of unknown cause, and found compound heterozygous KIAA0586 mutations in four additional unrelated patients with Joubert syndrome (table 2). Patient 3 was evaluated twice at ages 4.4 and 7.8 years (figure 1B, first panel, P3; table 1). He presented at 4 months with hypotonia and developmental delay. Brain MRI showed typical features of Joubert syndrome (figure 1B 1,2); cerebral cortex was unremarkable (figure 1B 3). At age 4.4, his chest size was normal, but his height was below average (height Z score was −2.06) (table 1). He had global developmental delay. Expressive speech was the most severely affected; he remained non-verbal at age 7.8 years. Patient 3 had the same multiple exon deletion present in patient 1 and patient 2, in addition to a c.428delG; p.Arg143Lysfs*4 frameshifting deletion (KIAA0586: c.428delG; p.Arg143Lysfs*4) that leads to an early termination.
KIAA0586 mutations in our patients with Jeune and Joubert syndromes
Patient 4 (figure 1B, second panel, P4; table 1) had an unremarkable perinatal history. In early infancy, he required multiple hospital admissions due to respiratory distress in the context of a viral infection. He walked at 26 months, and had several words after 12 months, but had speech articulation problems. Brain MRI showed severely hypoplastic and dysplastic vermis (figure 1B 4 and 5) and normal cerebral cortex (figure 1B 6). Chest size and height were normal at age 4 years (figure 1B, second panel, P4; table 1). Eye examination showed unilateral form fruste retinal coloboma. Wechsler Intelligence test at NIH showed a full-scale IQ of 88.
Patient 5 (figure 1B, third panel, P5; table 1) had a prenatal ultrasonography at 15 weeks that showed a ‘cerebellar cyst’, and Dandy–Walker was suspected. Oligohydramnios was noted at 35 weeks. Postnatally, she did well without breathing or feeding problems. At 6 months, she was admitted due to respiratory distress during a viral respiratory infection. She was overweight at age 6 months; molecular genetic testing for Prader–Willi syndrome was negative. Eye examination at the NIH revealed bilateral lateral gaze palsy. Chest size was normal, but her height was below average (height Z score −2.01) (table 1).
Patient 6 (figure 1B, fourth panel, P6; table 1) was born at term. He had intermittent tachypnoea from birth to 2 years. Hypotonia and developmental delay were noted at 10 months. The cerebral cortex was normal on MRI (figure 1B 11). Beginning at age 10 years, he had four afebrile grand mal seizures responsive to carbamazepine. In addition, he had problems with behaviour, including difficulty with anger control. Eye examination showed normal acuity, but a moderately constricted visual field. Optic atrophy was noted on examination (see online supplementary figure 1B). The patient was cooperative with optical coherence tomography revealing thinning of the retinal nerve fibre layer consistent with a diagnosis of optic atrophy. Full scale IQ was 56 on Wechsler Intelligence test performed at NIH.
Patients 4, 5 and 6 had the same 1 bp deletion seen in patient 3 (table 2). In addition, patient 4 had c.1159C>T; p.Gln387* that leads to an early termination at amino acid 387. Patient 5 had c.1413-1G>C; this variant is predicted to lead to the skipping of exon 12 (Berkeley Drosophila Genome Project (BDGP), see URLs). Patient 6 had c.1120+1G>A that is predicted to lead to an insertion of 802 nucleotides (BDGP, see URLs) and likely leads to an early termination 5 amino acids after exon 9.
Expression of KIAA0586 in normal tissues and patient cells
KIAA0586 (Gene ID: 9786), also known as TALPID3, has several isoforms (figure 2B). The longest isoform (NM_001244189.1) consists of 34 exons, spans ∼121 kb and codes for a 1644 amino acid protein (figure 2C). In GenBank, five additional isoforms have been described. Interestingly, all our mutations are found before the highly conserved domain of KIAA0586 that is necessary for centrosome localisation (figure 2C). Although all seven different mutations identified in our cohort are potentially deleterious alleles, they do not affect all of the six known isoforms (see online supplementary table S2).
KIAA0586 is ubiquitously expressed in various tissues (figure 3A). Of note, however, is the difference in the levels of expression using two different probes that presumably amplify common areas in all known isoforms, suggesting that there may be other isoforms that have not yet been identified. We then looked at the effect of the mutations on the transcript level by measuring KIAA0586 mRNA transcripts of the three main coding isoforms, NM_014749.3, NM_001244189.1 and NM_001244190.1, using qPCR analysed with the comparative CT method. In fibroblasts of patient 1 (with Jeune–Joubert syndrome) and patient 3 (with Joubert syndrome), these three isoforms were markedly reduced compared with control (figure 3B), presumably due to nonsense-mediated decay. Interestingly, a greater reduction of KIAA0586 expression was seen in patient 1, indicating that this combination of variants is more severe or that the patients possess a modifier that affects KIAA0586 expression.
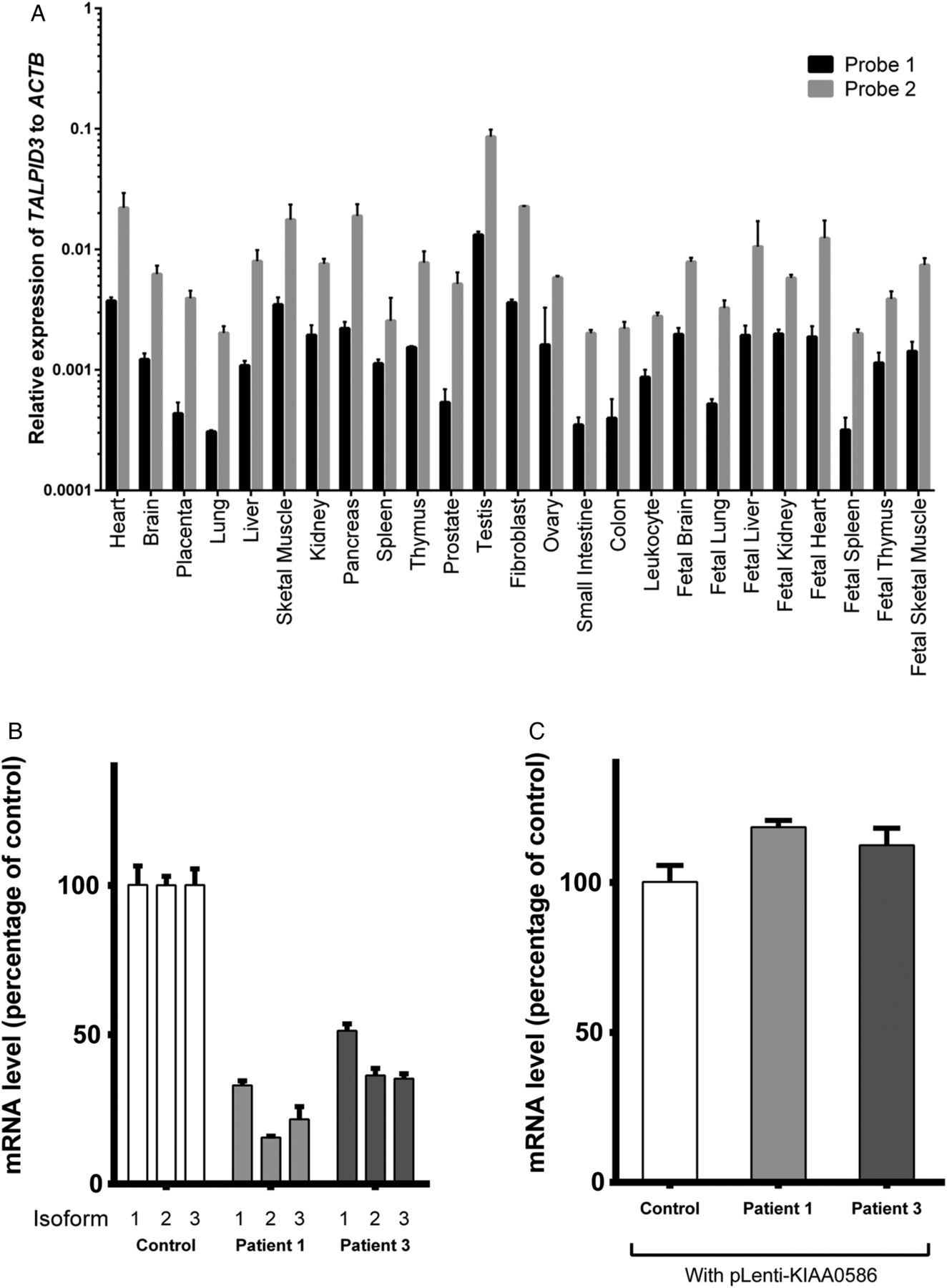
Expression of KIAA0586 in multiple tissue panel and cells. (A) Tissue expression of KIAA0586 in multiple tissue panels from normal, unaffected individuals using two probes that amplify the first two and the last few exons of the gene, respectively (See figure 2B for probe sites). KIAA0586 appears ubiquitously expressed, but the levels of expression vary with the two probes. (B) Analysis of common coding isoforms of KIAA0586 in affected individuals compared with unaffected control samples. mRNA was extracted from the fibroblasts of control individual (control), patient 1 (P1) and patient 3 (P3) and analysed by QPCR using primer pairs that specifically amplify the known coding isoforms of KIAA0586 (NM_014749.3, isoform 1; NM_001244189.1, isoform 2; and NM_001244190.1, isoform 3). Expression of all three isoforms was markedly reduced in P1 and reduced by half in P3. (C) Overexpression of the full length human KIAA0586 (NM_014749.3) in fibroblasts by lentiviral-mediated transduction led to recovery of expression in both P1 and P3. KIAA0586 isoform expression was normalised to that of POLR2A.
Patients with KIAA0586 mutations show reduced cilia length in primary fibroblasts
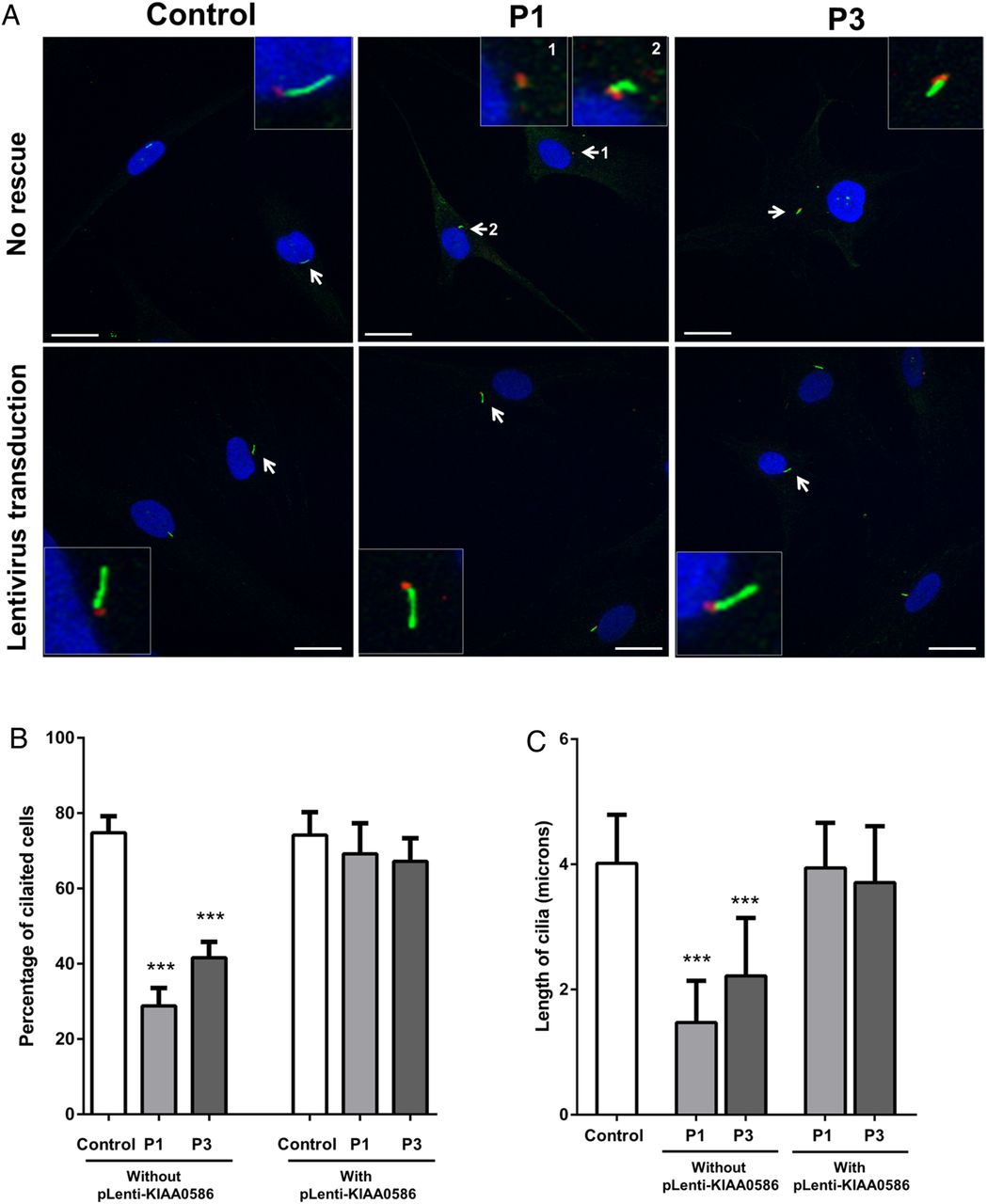
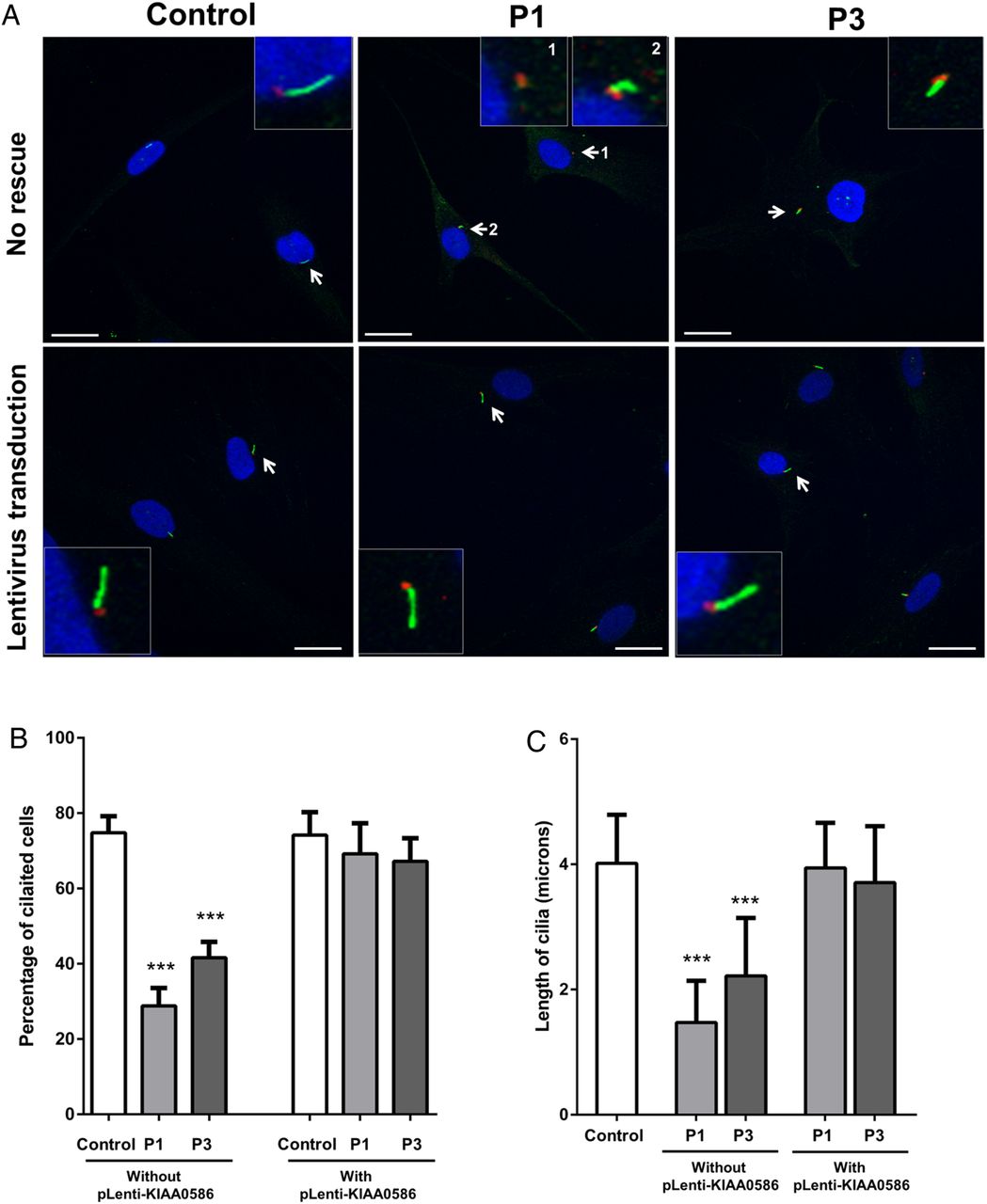
Therefore, using cilia-specific antibodies and immunofluorescence microscopy, we examined the ability of primary fibroblasts from patients 1 and 3 to form normal cilia. After 48 h of serum starvation, up to 80% of control cells were ciliated, while only 30% of patient-1 cells and 50% of patient 3 cells were ciliated (figure 4A). Cilia length was markedly reduced in both patients 1 and 3 (figure 4A). Transduction of cells with the full length KIAA0586 using lentiviral-mediated system rescued the number of ciliated cells and the cilia length in both patients 1 and 3 (figure 4B). The impaired cilia formation in the cells of patients 1 and 3 supports the prediction that the deleterious variants found in our cohort disrupt the well-conserved domain of KIAA0586 that is necessary for centriole assembly and, consequently, cilia formation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of cilia in patient 1 and patient 3 with mutations in KIAA0586. (A) Fibroblasts from control, patient 1 (P1) and patient 3 (P3) were grown to near confluence and serum starved to allow formation of cilia. Forty-eight hours after serum starvation, cells were fixed in methanol and probed with anti-ARL13B antibody (green) that marks cilia and mouse antiacetylated tubulin (red) that stains the basal bodies. Insets show magnified images of cells (arrows). (B) Approximately 80% of control cells have cilia, while only 30% and 50% of cells are ciliated in P1 and P3, respectively. Overexpression of the full length human KIAA0586 (NM_014749.3) in fibroblasts by lentiviral-mediated transduction increased the number of ciliated P1 and P3 cells to normal. Scale bars represent 20 µm. Asterisks indicate p value <0.001 between control and patient (P1 or P3) cells using non-parametric t test. (C) Cilia length in cells was measured using Zen 2009 software (Zeiss). In control cells, cilia were ∼3.5–7 μ in length. In P3, the cilia length ranged from 0.05 to 2.5 μ. In P3, cells had longer cilia than P1 (without pLenti-KIAA0586), measuring 2.5–5 μ, but still shorter than control. The length of cilia in P1 and P3 cells returned to normal after transduction with full length human KIAA0586 (with pLenti-KIAA0586). Asterisks indicate p value <0.001 between control and patient (P1 or P3) cells using non-parametric t test.
Discussion
KIAA0586 plays a key role in cilia assembly15 ,16 in both cellular and animal models.17 Mutant mouse18 and zebrafish17 embryos lacking Kiaa0586 do not have primary cilia, and like talpid3 mutant chicken embryos, have face and neural tube defects. Mice and zebrafish that lack Kiaa0586, however, also have defects in left-right asymmetry.17 ,18 KIAA0586 is a component of a CP110-containing protein complex important for centrosome and cilia function.19 KIAA0586 assembles a ring-like structure at the extreme distal end of centrioles. Ablation of TALPID3 results in an aberrant distribution of centriolar satellites involved in protein trafficking to centrosomes; it also causes ciliary vesicle formation reminiscent of loss of Cep290, another CP110-associated protein that is mutated in various human ciliopathies19 ,20 and leads to failure of centrosome migration.20 A similar failure of centrosome positioning and/or docking is seen in cells lacking function in other genes encoding basal body proteins, including OFD1 (oral-facialdigital syndrome 1)21 and MKS1 (Meckel syndrome 1).22
Although patients with Joubert with KIAA0586 mutations have not been reported with findings of Jeune syndrome,23 there are other precedents for the coexistence of clinical characteristics of Joubert and Jeune syndromes. The chicken talpid3 mutant displayed a small chest and developed extremely short limbs with increased number of digits, many of which were morphologically identical.1 In human beings, mutations in CSPP1, a spindle pole-associated protein like KIAA0586, involved in the centrosome, were recently identified in patients with Joubert syndrome with features of Jeune syndrome.24 ,25 Finally, the product of CEP120, mutated in some patients with Jeune syndrome,23 interacts with KIAA0586.26 In mice, the localisation of Cep120 to daughter centrioles27 is dependent on Talpid3.26 This interaction may explain why the phenotypes observed in our patients ranged from Jeune to Joubert syndrome.
Similar to the findings of Alby et al,28 which were published while our paper was under review, our patients with KIAA0586 mutations show reduced number of ciliated cells in fibroblasts. It is important, however, to note that the effect on cilia formation is more profound in patient 1, whose expression of KIAA0586 at the RNA level is much more reduced as compared with patient 3. Measurement of the KIAA0586 protein levels may help clarify the issue in the future. In addition to the lack of cilia formation, we have also shown that the length of cilia in our patients’ fibroblasts are much shorter when compared with control; this finding may only be specific to human fibroblasts, as TALPID3/Talpid3 deficiency in various animal models show absence of cilia formation.
Multiple deleterious alleles or variants that are predicted to produce premature stop codon have been identified in KIAA0586 and reported in various databases (see online supplementary table S3).28–30 However, a discrepancy arises between the high frequency of these deleterious alleles (estimated prevalence 1/3500 in Exome Variant Server and 1/30 000 in Exome Aggregation Consortium; see URLs), and the actual prevalence of Joubert syndrome, estimated at 1:100 000.6 This may be related to the existence of multiple isoforms of KIAA0586, or to embryonic lethality of biallelic mutations. Also of note is the difference in severity of symptoms between patients as exemplified by patient 1 who exhibited a more severe presentation than other subjects. This again could be explained by the profound reduction in KIAA0586 expression in cells, or the differential effect of mutations in the various KIAA0586 isoforms. This may also explain why the phenotypes of patients with mutations in KIAA0586 reported to this date appear to have a wide range of severity.28–30 Further studies will be required to determine the effect of KIAA0586 variants on the different isoforms.
Our results confirm that KIAA0586/TALPID3 is essential in cilia formation in human beings, expand the KIAA0586 phenotype to include features of Jeune syndrome and provide a pathogenetic connection between Joubert and Jeune syndromes, based on aberrant ciliogenesis.
URLs
Exome Aggregation Consortium (http://exac.broadinstitute.org)
Exome Variant Server (http://evs.gs.washington.edu/EVS/)
dbSNP (http://www.ncbi.nlm.nih.gov/SNP/)
GenBank (http://www.ncbi.nlm.nih.gov/genbank/)
Acknowledgments
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute, National Eye Institute and the National Institute of Mental Health of the National Institutes of Health, Bethesda, Maryland, USA. We also thank the patients and their families for participating in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
MCVM, TV, JSt, WAG and MG-A contributed equally.
Contributors MCVM performed most cell biology experiments, provided direction for the project, analysed the data, generated figures and wrote the manuscript. TV performed most molecular data experiments, analysed the data, generated figures and contributed to manuscript writing. JSt helped in molecular data experiments, analysed the data, helped generate figures and contributed to manuscript writing. DM contributed to cell culture experiments and data analysis and manuscript writing. LM contributed to cell culture experiments and data imaging and analysis and manuscript writing. DK contributed to analysis of cilia in cells and manuscript writing. JG contributed to cloning experiments, production of lentivirus and manuscript writing. DY assisted in screening for mutations in our ciliopathy cohort and contributed to manuscript writing. JB coordinated admitting the patients, gathering data from patients and contributed to manuscript writing. RF assisted in screening for mutations in our ciliopathy cohort and contributed to manuscript writing. WMZ gathered and analysed ophthalmological data from patients and contributed to manuscript writing. JSn gathered and analysed neurocognitive data from patients and contributed to manuscript writing. MV contributed to tools for data analysis and manuscript writing. JCM contributed to tools for data analysis and manuscript writing. CT gathered and analysed neurological data from patients and contributed to project direction and manuscript writing. BDS contributed to tools for data analysis and manuscript writing. JEN contributed to tools for data analysis and manuscript writing. NISC Comparative Sequencing Program performed whole exome sequencing and tools for data analysis. WAG provided direction for the project, obtained funding, guided data collection and interpretation and contributed to manuscript writing. MG-A, principal investigator of clinical trial NCT00068224, designed research, recruited patients, performed clinical and molecular data analysis, provided direction for the project and wrote the manuscript.
Funding This research was supported by the Intramural Research Programs of the National Human Genome Research Institute, National Eye Institute and the National Institute of Mental Health of the National Institutes of Health, Bethesda, Maryland, USA.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Institutional Review Board of the National Human Genome Research Institute, National Institutes of Health.
Provenance and peer review Not commissioned; externally peer reviewed.