Article Text

Abstract
Background CBL missense mutations have recently been associated with juvenile myelomonocytic leukaemia (JMML), an aggressive myeloproliferative and myelodysplastic neoplasm of early childhood characterised by excessive macrophage/monocyte proliferation. CBL, an E3 ubiquitin ligase and a multi-adaptor protein, controls proliferative signalling networks by downregulating the growth factor receptor signalling cascades in various cell types.
Methods and results CBL mutations were screened in 65 patients with JMML. A homozygous mutation of CBL was found in leukaemic cells of 4/65 (6%) patients. In all cases, copy neutral loss of heterozygosity of the 11q23 chromosomal region, encompassing the CBL locus, was demonstrated. Three of these four patients displayed additional features suggestive of an underlying developmental condition. A heterozygous germline CBL p.Y371H substitution was found in each of them and was inherited from the father in one patient. The germline mutation represents the first hit, with somatic loss of heterozygosity being the second hit positively selected in JMML cells. The three patients display a variable combination of dysmorphic features, hyperpigmented skin lesions and microcephaly that enable a ‘CBL syndrome’ to be tentatively delineated. Learning difficulties and postnatal growth retardation may be part of the phenotype.
Conclusion A report of germline mutations of CBL in three patients with JMML is presented here, confirming the existence of an unreported inheritable condition associated with a predisposition to JMML.
- JMML
- CBL
- microcephaly
- cancer predisposition
- molecular genetics
- haematology (incl blood transfusion)
- paediatric oncology
Statistics from Altmetric.com
- JMML
- CBL
- microcephaly
- cancer predisposition
- molecular genetics
- haematology (incl blood transfusion)
- paediatric oncology
Introduction
The Casitas B-cell lymphoma (CBL, c-CBL) protein is a member of the CBL family of E3 ubiquitin ligases. CBL controls proliferative signalling networks by downregulating the growth factor receptor signalling cascades.1 2 CBL contains a tyrosine kinase binding (TKB) domain and a zinc binding RING finger domain that mediates the E3 ubiquitin ligase activity. These two highly conserved domains are separated by a short linker sequence crucial for ubiquitin ligase activity of CBL.1 The E3 ligase activity directs the mono-ubiquitination of activated receptors at multiple sites, which promotes endocytosis and lysosomal degradation of the receptors.2 CBL is also involved in many signalling events through its function as a multiadaptor protein.
CBL missense mutations have recently been associated with acute myeloid leukaemia,3 4 various myeloproliferative neoplasms,5 6 and juvenile myelomonocytic leukaemia (JMML).7 JMML is an aggressive myelodysplastic and myeloproliferative neoplasm of early childhood characterised by excessive macrophage/monocyte proliferation that infiltrates haematopoietic and non-haematopoietic tissues.8 The natural course of JMML is rapidly fatal. Progression to acute leukaemia is infrequent, but most children die from progressive respiratory and multivisceral failure. Allogenic bone marrow transplantation is the only curative therapy, achieving long term survival in about half of the patients. Cells from affected patients are abnormally sensitive to granulocyte–macrophage colony stimulating factor (GM-CSF). This hypersensitivity is the result of the pathological activation of the RAS signalling pathway by mutations of NRAS, KRAS, NF1, PTPN11 or CBL.7 8 In JMML, CBL mutations were found to target preferentially amino acid Y371 in the linker region, whereas mutations affecting this amino acid are rare in other myeloid neoplasms. As in other myeloid malignancies, CBL mutations found in JMML are associated with loss of the non-mutated CBL allele by acquired uniparental disomy (UPD) of the 11q23 chromosomal region.5 9
JMML has been observed in association with congenital malformations, including neurofibromatosis type 1 (NF1) (OMIM 162200) and Noonan syndrome (NS) (OMIM 163950), two conditions associated with RAS pathway deregulation.8 Patients with NF1 display loss of the normal NF1 allele in cancer cells.10 Patient with NS and JMML usually harbour activating heterozygous germline mutations in PTPN1111 and, more rarely, in KRAS12 or NRAS.13
Germline CBL mutation was suspected in one patient by Loh et al7 who detected a heterozygous p.Y371H CBL mutation in cord blood and a homozygous p.Y371H mutation in JMML cells. We confirm here that some patients with JMML can harbour germline mutations of CBL by reporting three further patients among a cohort of 65 JMML patients. Our observations suggest that constitutional CBL mutations lead to a constellation of mild, variable developmental defects and predisposition to JMML, which could be tentatively described as a ‘CBL syndrome’.
Patients and methods
Patient 1
General history
Patient 1 was born at 38 weeks of gestation (WG). Her mother had gestational diabetes. Birth weight (BW) was 3020 g (−0.6 SD), birth length (BL) 45 cm (−2.4 SD), occipitofrontal head circumference (OFC) 32 cm (−2.2 SD). She had failure to thrive, with poor sucking and postnatal growth retardation. She walked at the age of 24 months. She uttered her first words at 23 months. Subsequent development of speech was severely delayed. She had normal hearing and normal ophthalmologic examination. She developed JMML at age 26 months. At first evaluation, in the genetic department of Nantes, at age 30 months, she was 78 cm tall (−3 SD), had a weight of 9.6 kg (−2.1 SD), and an OFC of 44.5 cm (−2.7 SD). She was felt to be mildly dysmorphic, disclosing broad forehead, hypertelorism, epicanthic folds, deeply grooved philtrum, thick lips, mild retrognathism, thick, posteriorly rotated but normally set ears with overfolded helices, short neck, thin hair and low posterior hairline (figure 1A, B). She had a single café-au-lait spot on the abdomen. She had no thoracic or spinal deformities. The cardiac ultrasound scan was normal. The cerebral magnetic resonance image (MRI) was normal. She was hyperactive, with short attention span and poor verbal skills. At that time, a diagnosis of mild Noonan syndrome was tentatively suggested. At last evaluation, aged 4 years 6 months, her height was 96 cm (−1.7 SD), her weight was 13.7 kg (−1.3 SD), and her OFC was 47 cm (−2.2 SD).

Photographs of the three patients. A, C, E: frontal facial view of patient 1 (at 4 years 6 months of age), of patient 2 (at 24 months of age), of patient 3 (at 6 years of age). B, D, F: lateral facial view of patient 1 (at 30 months of age), of patient 2 (at 24 months of age), of patient 3 (at 6 years of age).
Family history
This girl was the second child of healthy non-consanguineous parents aged 27 years and 22 years at time of her birth. The father was 180 cm tall (+0.8 SD). He has no personal or familial history of haematologic malignancies. He had a vocational training qualification. The mother was 155 cm (−1.5 SD). Several relatives of the mother had adult onset cancers of various types.
Haematological history
At age 26 months, she was hospitalised for massive hepatosplenomegaly, retroperitoneal and mesenteric lymphadenopathies, hyperleucocytosis (46×109/l), thrombocytopenia (46×109/l) and anaemia (8.4 g/dl). The peripheral differential blood count showed: 17% monocytes (7.6×109/l), 18% immature granulocytes, and 3% blasts. The bone marrow aspirate was hypercellular with granulocytic proliferation but without excess of blasts. These features were consistent with a diagnosis of JMML. She received a cord blood allograft, then relapsed and benefited from a second one. She is alive, 16 months after the second allograft and displays complete donor chimerism.
Patient 2
General history
Patient 2 was born in Tunisia at 41 WG by caesarean section for dystocia. Her BW was 3100 g (−0.4 SD), BL 50 cm (−0.9 SD), and OFC 34 cm (−1 SD). The pregnancy was uneventful. She had postnatal failure to thrive. At age 1 year, her height was 71 cm (−0.6 SD), her weight was 8 kg (−1.2 SD), and her OFC was 44 cm (−1.1 SD). At last evaluation, at age 24 months she weighed 8 kg (−3 SD), her height was 77 cm (−2.4 SD), and her OFC was 44.5 cm (−2.4 SD). She had microcephaly, triangular facies, high cranial vault, bilateral epicanthic folds, thick lips, prominent philtrum, posteriorly rotated helices, and somewhat sparse hair (figure 1C, D). The echocardiogram was normal. She has no skin anomalies. T2 weighted fluid attenuated inversed recovery (FLAIR) MRI showed non-specific hyperintense signals in the periventricular white matter. Neurological examination was normal. Psychomotor development was appropriate for age (walked at 18 months, first words at 12 months). Bone age was mildly delayed (18 months at age 22 months). IGF1 was 12 nmol/l (normal range (NR) 4–22 nmol/l) and IGFBP-3 1100 ng/ml (NR 1090–2490 ng/ml).
Family history
She was the only child from healthy first cousins parents. The mother was 26 years old and the father 39 years old at birth. The father was 168 cm tall (−0.6 SD) and the mother 160 cm (−1.2 SD). The family history was not contributory.
Haematological history
A diagnosis of JMML was made at 13 months of age because of persistent hyperleucocytosis (50×109/l), thrombocytopenia (35.109/l), and hepatosplenomegaly. The peripheral differential blood count showed 19% monocytes (9.5×109/l), 9% immature granulocytes, and 0.5% blasts. The bone marrow was hypercellular with granulocytic proliferation and no excess of blasts. At age 24 months, she received a cord blood allograft. She remains alive at age 25 months. A mixed chimerism (50% donor) was found 30 days after the allograft.
Patient 3
General history
Patient 3 was born at 39 WG. Her mother had gestational diabetes. BW was 3240 g (−0.1 SD), BL 50 cm (+0.3 SD), and OFC 35 cm (+0.55 SD). At age 12 months, she was 70.5 cm tall (−0.8 SD), and weighed 8.8 kg (−0.4 SD), and her OFC was 45 cm (−0.25 SD). She had a broad forehead, arched eyebrows, hypertelorism, palpebral ptosis, short, upturned nose, flat malar areas, deeply grooved philtrum, posteriorly rotated but normally set ears with thick helices and large lobules (figure 1E, F). Pectus excavatum and hypermobile finger joints were present. Her skin was hyperelastic and she presented redundant skinfolds on hands and feet. Three café-au-lait spots (<2 cm) were present on her back and two were on the anterior part of the thighs. The cardiac ultrasound and cerebral MRI were normal. Neurological and ophthalmologic examinations and hearing test were normal. She walked unsupported at age 14 months and uttered her first words at 18 months. She displayed a postnatal decline of her OFC (at age 2 years her OFC was 46 cm (−1.2 SD) and at age 3 years 47.5 cm (−1.1 SD)). At last evaluation at the age of 6 years, her height was 107 cm (−1.4 SD) and her weight was 15 kg (−1.9 SD). She had normal schooling (first grade) and no learning disabilities.
Family history
This girl was the only child of healthy non-consanguineous parents aged 27 years and 23 years at the time of her birth. The father was 193 cm tall (+3.0 SD) and the mother 153 cm (−1.8 SD). The family history was not contributory.
Haematological history
At age 12 months, she was hospitalised for massive splenomegaly, hyperleucocytosis (30×109/l), mild thrombocytopenia (136×109/l), and anaemia (9.4 g/dl). The peripheral differential blood count showed: 22% monocytes (6.6×109/l), 5% immature granulocytes, and 1% blasts. The bone marrow aspirate was hypercellular with granulocytic proliferation but without excess of blasts. She had a cord blood allograft. She had no relapse 4 years and 9 months after the allograft. She has complete donor chimerism.
Genetic analysis
Parents gave written consent for genetic analysis and clinical photographs.
Karyotyping
Conventional cytogenetic analysis was performed on peripheral blood and bone marrow using standard procedures.
Mutation screening
Bone marrow aspirates and peripheral blood were collected on EDTA at diagnosis. Genomic DNA was extracted from mononucleated haematopoietic cells, fibroblasts or buccal swabs using Qiagen Mini or Midi Kit (Qiagen, Valencia, CA, USA). Mutation screening was performed by bidirectional sequencing of exons and their flanking intron–exon boundaries as described previously.14 The entire coding region of KRAS and NRAS was screened. PTPN11 screening was restricted to exons 2, 3, 4, 7, 8, 12, 13, 14 and CBL screening to exons 7, 8 and 9. GenBank accession number for CBL genomic and mRNA reference sequences are NM_005188 and NC_000011, respectively.
Microsatellite analysis
Loss of heterozygosity (LOH) at CBL locus was assessed by PCR amplification of nine microsatellite markers covering the 11q arm: D11S1294 (11q22.3), D11S4206 (11q22.3), D11S4129 (11q22.3), D11S924 (11q22.3), D11S4171 (11q22.3), D11S1774 (11q22.3), D11S925 (11q23), D11S934 (11q23-24), D11S968 (11q25).
LOH at NF1 locus was assessed by PCR amplification of eight microsatellite markers covering the 17q arm located within or close to the NF1 gene, respectively.15
Family links were checked by testing a panel of 16 microsatellite using the Powerplex kit (Promega, Madison, WI, USA).
Single nucleotide polymorphism (SNP) array analysis
Tumour (bone marrow cells) and fibroblasts DNA were hybridised to Affymetrix Genome-Wide Human SNP 6.0 Arrays. CEL files were created using the Affymetrix GeneChip Command Console operating software and Genotyping Console 2.1, according to the manufacturer's protocols (Affymetrix, Santa Clara, CA, USA). The Partek Genomics Suite was used for both copy number alteration (CNA) and LOH analysis. Regions of CNA were detected using a Hidden Markov Model algorithm in the standard Partek workflow for paired samples. LOH was assessed by comparing paired germinal and tumour samples using Partek LOH workflow.
Myeloid progenitor cell growth
In vitro growth of myeloid progenitors was performed by plating bone marrow and peripheral blood mononucleated cells in semi-solid methylcellulose with and without leucocyte conditioned medium (cytokines medium, LCM, StemCell Technologies Inc, Vancouver, Canada) as described previously.16 Colonies (aggregates containing >50 cells) were scored at day 11 and 14. ‘Endogenous growth’ corresponded to the presence of colony forming units (CFU) (CFU-GM >10 colonies or CFU-M >5 colonies) in absence of growth factors.
Results
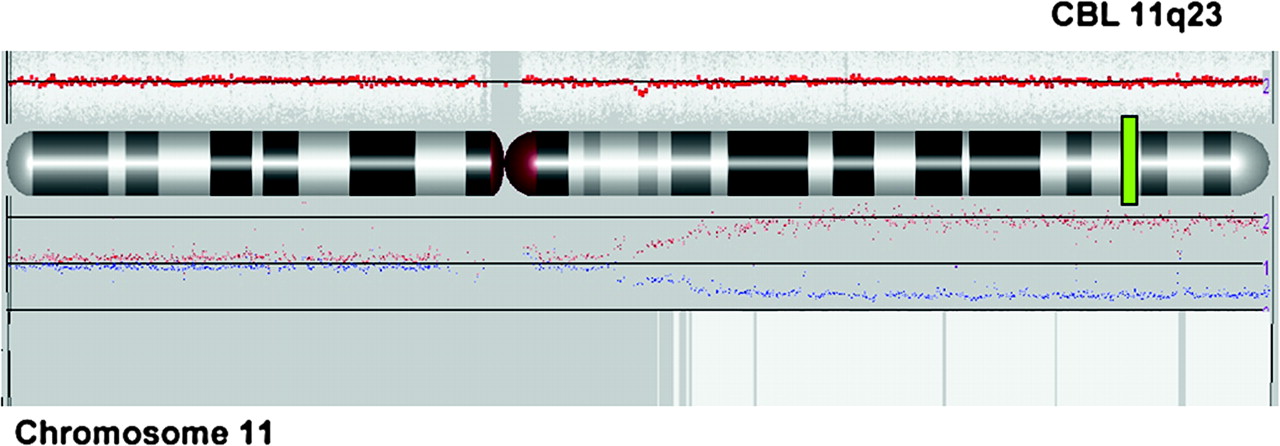
CBL mutations were screened in a cohort of 65 unselected patients with JMML. A homozygous mutation of CBL was found in leukaemia cells of 4/65 (6%) patients. In all patients, copy neutral loss of heterozygosity of the 11q23 chromosomal region, encompassing the CBL locus, was demonstrated by SNP array (figure 2) and microsatellite analysis. No RAS activating mutation in NRAS, KRAS, or PTPN11 gene (classically associated with JMML) was detected. None of the patients had phenotypic feature evocative of NF1, except for some café-au-lait spots in patients 1 and 3. Mitotic recombination leading to loss of heterozygosity and subsequent inactivation of both NF1 alleles is virtually constant in JMML of patients with neurofibromatosis.10 No loss of heterozygosity at the NF1 locus was present in the leukaemia cells of our three patients, making unlikely the presence of a NF1 in these children.
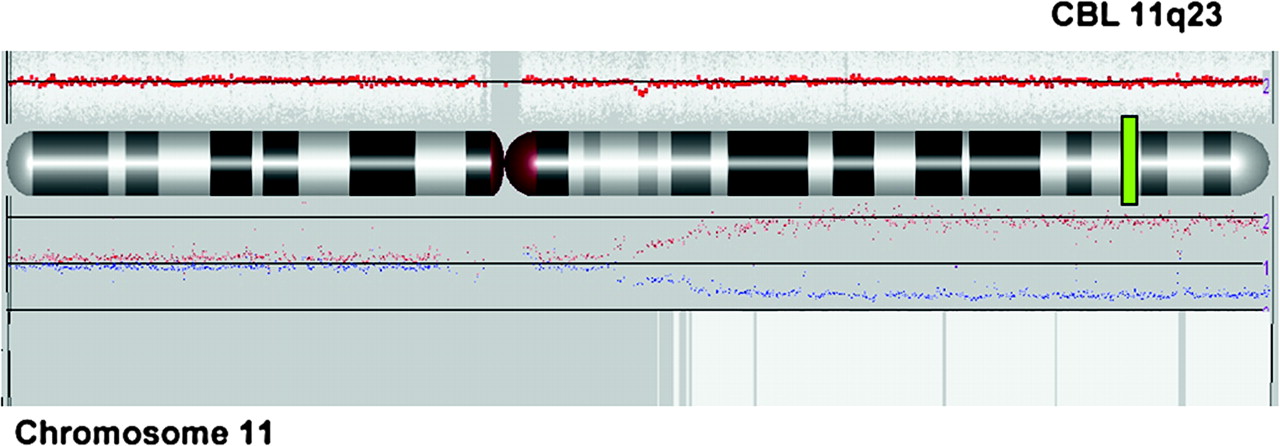
Single nucleotide polymorphism (SNP) array profile of chromosome 11 (Affymetrix SNP6.0 technology and Partek image) showing the copy neutral loss of heterozygosity (LOH) of tumour versus germinal DNA for patient 1. The red line represents average copy number (CN) signal intensity of SNPs on the array chip. In this instance, there are no CN variations and the red line does not deviate from normal diploid CN. The graph above chromosome ideogram represents the allele specific copy number and indicates copy neutral LOH. White box represents uniparental disomy (UPD) region.
One JMML patient had a c.1254C→G homozygous transversion in exon 9 of CBL, resulting in the p.F418L missense amino acid substitution. The three others had a c.1111T→C homozygous transition (in exon 8), resulting in the p.Y371H missense amino acid substitution (figure 3.1). These patients displayed additional features suggestive of an underlying developmental condition. All three had normal blood and bone marrow karyotype. A heterozygous germline p.Y371H substitution was found in fibroblasts in each of them (figure 3.2). DNA of cheek mucosa (obtained from buccal swabs) was tested in patients 2 and 3 and also showed the presence of the germline p.Y371H substitution. In patient 1, despite a family history of cancer in maternal lineage, the mutation was inherited from the father (figure 3.4). Mutation occurred de novo in the two other patients (figure 3.4).

{kind=link}
{kind=link}
{kind=link}
Germline CBL mutations. Sequence electropherograms documenting the c.1111T→C mutation found in exon 8 of CBL gene for these three patients: 3.1, DNA at time of the leukaemia diagnosis; 3.2, DNA from cultured fibroblasts; 3.3, maternal DNA; 3.4, paternal DNA. Asterisks indicate homozygous state; crosses indicate heterozygous state.
These three patients fulfilled all the JMML diagnosis criteria reported at the last JMML international symposium17 and their presentation did not significantly differ from that observed in the other patients (age at diagnosis, white blood cell count, etc). Myeloid progenitors from blood and bone marrow showed an endogenous growth pattern in the absence of growth factors, as classically observed in JMML (data not shown).
The three children present slightly dysmorphic traits. Two patients have café-au-lait spots. Two have microcephaly below −2 SD. Patient 1 had short birth length, and secondary catch up ending in low normal stature at age 4.5 years. Patient 2 had true growth decline during her second year of life, ending with true postnatal growth retardation. No growth retardation was noted in patient 3 and in the father of patient 1.
Discussion
We identified constitutional heterozygous missense mutations of CBL in three unrelated patients with JMML, confirming the existence of a currently unrecognised dominantly inherited condition with propensity to develop myeloproliferative neoplasm (JMML) in early infancy. Our patients carried the recurrent p.Y371H substitution, inherited from the father in one instance. The process of tumorigenesis observed in our patients is in line with the classical Knudson hypothesis for tumour suppressor genes: the first hit is germline while the second hit occurs somatically and is selected for in JMML cells. Phosphorylation of Y371 is essential for the E3 activity of CBL and for its interaction with a number of signalling proteins. Phospho-Tyr-371 appears to play a role in the interactions of c-Cbl with PI3 kinase (PI3K).2 Substitution of Y371 can have different consequences: mutants of CBL lacking Y371 are oncogenic,18 Y371F mutants are inactive in auto-ubiquitylation assays in vitro, while Y371E mutants apparently mimic phosphorylated tyrosines and are constitutively active.2
Although our series is small, we found subtle developmental anomalies in all patients. Mild hypertelorism, short upturned nose, deeply grooved philtrum and thick lips, reminiscent of the facial gestalt of Noonan syndrome, are observed in the three patients. Two patients have café-au-lait spots. Patient 1 had short stature at birth and low normal height at last evaluation, whereas patient 2 showed progressive growth retardation. These two children have mild microcephaly, present at birth in one of them. In patient 1, developmental milestones are delayed. For patient 2, there is no major developmental anomaly, but her young age precludes any firm conclusion. Patient 3 had a normal head circumference at birth and presented a postnatal decline of her OFC. She had a normal development, as did patient 1's father. Although these anomalies could have occurred by chance, presence of some dysmorphic signs in each patient led us to raise the hypothesis that CBL haploinsufficiency may interfere with normal somatic and cerebral development. Unfortunately, no phenotypic data are available for the only other patient with constitutional CBL mutation reported by Loh et al.7 At this point, these somewhat conflicting observations do not allow us to make a firm conclusion, considering the possible confounding effects of JMML development itself, and of therapeutic and nutritional interventions. The dysmorphic traits are subtle, and need to be confirmed by independent reports and long term follow-up, but the fact that Noonan syndrome was discussed in case 1 before the discovery of the CBL anomaly has to be underlined. Time will tell if our seminal observations represent the first step in the delineation of a ‘CBL syndrome’.
RAS activation is known to play a central role in sporadic and syndromic JMML. CBL germline mutations are associated with a propensity to develop JMML, suggesting that they induce RAS pathway activation. However, they are associated with a phenotype that is distinct from Noonan syndrome, neurofibromatosis, or any other disorder belonging to the spectrum of RAS-mitogen activated protein kinase (MAPK) activation syndrome. Significantly, congenital heart defects were not observed in our patients.
Abnormalities in the ubiquitylation system have been implicated in the pathogenesis of various human disease including malignant transformation and several genetic diseases.19 Knockout of either the Cbl or Cbl-b genes in mice are mainly characterised by a phenotype in the lymphocyte compartment.20 21 However, Cbl−/− mice also display a relatively mild phenotype in other cell types. In addition, recent data suggest that mutations do not lead to the simple knockout of Cbl function but render it a proto-oncogene.22 Mutant CBL inhibits ubiquitination of growth factor receptors even in the presence of a normal copy of the CBL gene, leading to enhanced proliferative response to growth factors.6 22 Altogether, these observations are consistent with a phenotypic expression of CBL heterozygous mutation.
The importance of Cbl in haematopoiesis has been demonstrated in knockout mice that show hyperresponsiveness to haematopoietic growth factors, expansion of the progenitor and stem cell pool, and mild myeloproliferative features.22 The observation of a propensity to develop JMML in children with CBL germline mutations as well as the close association of CBL mutations with other monocyte expansions, such as that seen in chronic myelomonocytic leukaemia (CMML) or in acute myeloid leukaemia (AML) with monocytoid features, suggest a primary role for CBL mutations in the pathogenesis of these diseases. The reason for the specific effect of CBL on the monocytic lineage and the role of RAS in this process has to be clarified.
Intriguingly, Chiusaroli et al reported that growth is transiently delayed in Cbl−/− mice.23 This could be explained by a delayed replacement of cartilage by bone as a consequence of a decreased motility of osteoclasts. CBL inactivation may also interfere with growth hormone (GH) signalling.24
How can we understand the presence of microcephaly in 2/3 patients? Dysregulation of neuroepithelial progenitors proliferation and survival is suspected to be a major cause of primary microcephaly.25 Loss of CBL function, by contributing to the activation of the JNK pathway, may sensitise healthy neuronal cells to death.26 It has been recently shown that deficiency of the small GTPase Rac-1 in mouse forebrain causes microcephaly by increasing apoptosis and defective differentiation.27 Thus, decreased activation of RAC-1 by CBL may also play a role by inducing a pro-death signalling cascade.2
In conclusion, presence of germline alteration of CBL yields a relatively mild phenotype in childhood, possibly because of partial redundancy of CBL and CBL-b functions. However, it raises several concerns for the follow-up of those patients. Inappropriate activation of mammalian protein tyrosine kinases can lead to various forms of human cancers. Could CBL mutation predispose to other malignancies, or to secondary solid tumours in JMML survivors? Which prevention strategy should be proposed?
Acknowledgments
We thank the families for active participation in the study and for providing us with written consent for publication of their clinical and molecular data. We thank B Leheup for his advice in patient 3 and RA Padua for reviewing the manuscript. SNP-array analyses have been performed in the ‘Plateforme de Génomique du GHU Nord’ (J Soulier, S Quentin, P Luizzi). This work was supported in part by a grant from Enfant & Santé and the Société Française de lutte contre les Cancers et les leucémies de l'Enfant et l'adolescent (SFCE).
References
Footnotes
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.