Article Text

Abstract
Dyggve-Melchior-Clausen syndrome (DMC) is an autosomal recessive condition characterised by short trunk dwarfism, scoliosis, microcephaly, coarse facies, mental retardation, and characteristic radiological features. X rays show platyspondyly with double vertebral hump, epiphyseal dysplasia, irregular metaphyses, and a characteristic lacy appearance of the iliac crests. Electron microscopy of chondrocytes have shown widened cisternae of rough endoplasmic reticulum and biochemical analyses have shown accumulation of glucosaminoglycan in cartilage, but the pathogenesis of DMC remains unexplained. Here, we report on the homozygosity mapping of a DMC gene to chromosome 18q21.1 in seven inbred families (Zmax=9.65 at θ=0 at locus D18S1126) in the genetic interval (1.8 cM) defined by loci D18S455 and D18S363. Despite the various geographical origins of the families reported here (Morocco, Tunisia, Portugal, and Lebanon), this condition was genetically homogeneous in our series. Continuing studies will hopefully lead to the identification of the disease causing gene.
- Dyggve-Melchior-Clausen disease
- homozygosity mapping
- chromosome 18q21.1
Statistics from Altmetric.com
Dyggve-Melchior-Clausen syndrome (DMC, MIM 223800) is a rare autosomal recessive condition first described in 1962 by Dyggve et al1 and further studied by Naffah.2 The patients present with severe progressive short stature(<-5 SD), microcephaly, and facial dysmorphism. Clinical features include coarse facies with bulky jaws, short trunk, scoliosis, proximal limb shortening, broad hands and feet, and mental retardation.3,4 Radiological features include platyspondyly with double vertebral hump, small iliac wings with lacy iliac crest, dysplastic acetabulum, epiphyseal dysplasia, short long bones with irregular metaphyses, and short hands.5 This condition shares several clinical features with Morquio’s disease (MPS IV6) but, in contrast with MPS IV, no corneal clouding, deafness, valvular disease, or mucopolysacchariduria are observed, while microcephaly and mental retardation are consistently present in DMC. Biochemical and histochemical analyses have shown increased amount of keratan sulphate in cartilage and fibrous resting cartilage with markedly vacuolated chondrocytes and cytoplasmic inclusions.7,8 Electron microscopy has shown wide cisternae of rough endoplasmic reticulum in chondrocytes,7 but the catabolism of proteoglycans in DMC fibroblasts was reportedly normal.9 An inherited metabolic disorder has long been considered in DMC,10 but the disease mechanism and the disease causing gene(s) had remained unknown.
Here, we report on genetic linkage analyses in seven inbred families from Morocco (5/7), Tunisia (1/7), and Portugal (1/7) and in two non-inbred families from Morocco and Lebanon (fig 1). All affected subjects fulfilled the criteria for DMC, namely (1) severe short stature, (2) microcephaly, (3) short trunk with scoliosis and proximal limb shortening, (4) mental retardation, and (5) characteristic x ray manifestations, that is, double hump, centrally indented vertebral bodies, and lacy pelvis (fig 2). No abnormal urinary keratosulphate excretion was found in 5/9 families (table 1).
Clinical and radiological features in nine DMC families

Pedigrees and haplotypes of nine DMC families.

(A) Pictures of patients II.2 and II.3 (family 7) at 8 and 4 years. Note the rhizomelic dwarfism with short trunk, scoliosis, and coarse facies. (B) Pelvis and hip joints of patient II.2. Note the characteristic lacy appearance of iliac crest, the deformities of the femoral epiphyses, and the metaphyseal irregularities. (C) Spine of patient II.2. Note the double vertebral hump.
METHODS AND RESULTS
Informed consent and blood samples were obtained from all family members. Genomic DNA was purified from peripheral blood leucocytes according to standard techniques. Microsatellite DNA markers from the entire genome were used at an average spacing of 10 cM. For regions of interest, all samples were analysed with extra markers of the Genethon map using a single set of primers in each amplification reaction. The homozygosity mapping strategy was based on the assumption that affected subjects of the same kindred are homozygous by descent for the disease causing mutation11 and two point linkage analyses were performed using the MLink option of the LINKAGE package version 5.1.12 The disease gene frequency was set to 0.001 assuming an autosomal recessive mode of inheritance with complete penetrance. We took into account the inbreeding loops but allele frequencies were not available in the populations studied.
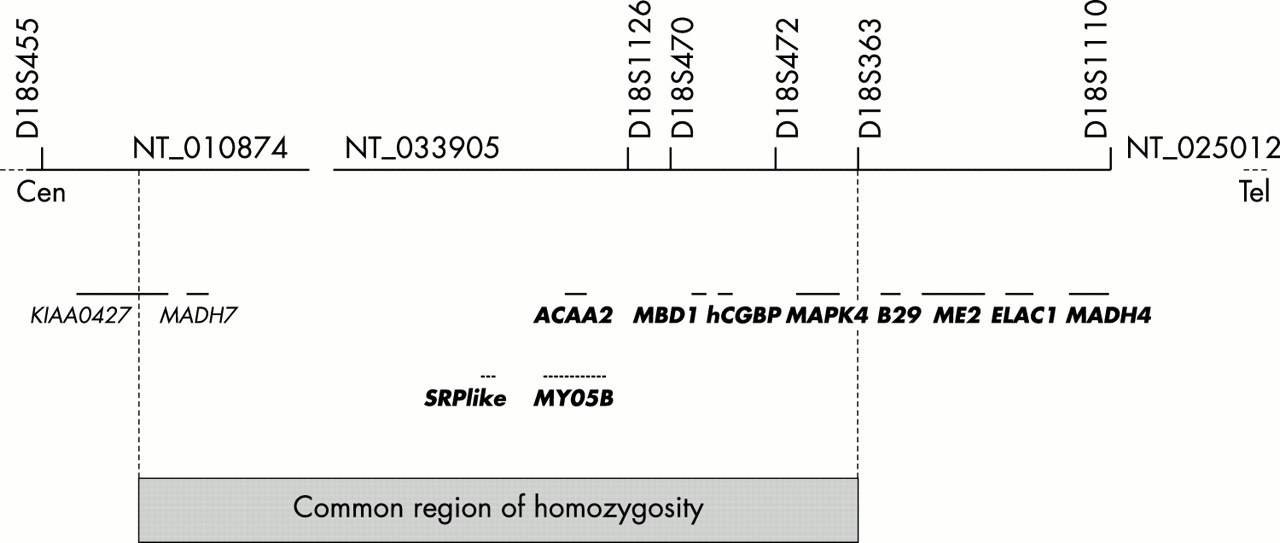
Six affected subjects from four families were initially tested (families 1-4) and four of six were homozygous for a marker at locus D18S363. Eleven additional markers were then tested in all nine families and we found linkage of the disease gene to polymorphic markers of chromosome 18q21.1. Pairwise linkage between a polymorphic marker at locus D18S1126 and the disease locus gave a maximum lod score of Z=9.65 at θ=0 (table 2). Heterozygosity at locus D18S455 in affected subjects II.1-3 (family 1) and II.1 (family 4) indicated that a recombination event had occurred between loci D18S455 and D18S1126, thus defining the proximal boundary of the genetic interval. Similarly, heterozygosity at locus D18S363 in affected subjects II.1, II.4, and II.5 (family 3) defined the distal boundary of the genetic interval encompassing the DMC gene (D18S455-D18S363, 1.8 cM, fig 3).
Pairwise lod scores for linkage of the DMC gene to chromosome 18q21.1

{kind=link}
{kind=link}
{kind=link}
Physical map of the region encompassing the DMC gene. Six genes, two predicted genes, and STS markers are indicated. The shaded region indicates the common region of homozygosity in the seven inbred families.
In this interval, we found heterozygosity for a CA repeat within KIAA0427 in affected subject II.1 (family 4), therefore reducing the shared region of homozygosity (fig 3).
DISCUSSION
Eight genes were considered as possible candidate genes by their position, namely the mothers-against-decapentaplegic-homologue 7 (MADH7), signal recognition particle 72 kD-like (SRP72-like), 3-oxoacyl-CoA thiolase (ACAA2), myosin Vb (MYO5B), MAP-kinase 4 (MAPK4), methyl-CpG binding domain protein 1 (MBD1) genes, and the human CpG binding protein gene (hCGBP, fig 3). Several of them are currently regarded as possible candidate genes by their function, namely ACAA2 which encodes a mitochondrial enzyme catalysing the last step of the mitochondrial fatty acid beta oxidation13 and MYO5B which belongs to the class V myosin family that function as motors for actin dependent organelle trafficking.14 Finally, we found that MBD1 and hCGBP are highly expressed in fetal brain, osteoblasts, and chondrocytes (data not shown). Whether any of these genes is involved in DMC is under investigation.
It is important to note that all DMC families but one were of Arab origin in our series. Whether the DMC gene frequency is high in the Middle East and North Africa or the DMC mutation confers a genetic advantage remains open to discussion.
In conclusion, we show here that a DMC gene maps to chromosome 18q21.1. Our results are consistent with genetic homogeneity of this condition. Continuing studies will help to decide whether Smith-McCort syndrome, which causes the same skeletal manifestations but no mental retardation,15 also maps to this region. Identifying the DMC gene will hopefully help to elucidate the pathogenesis of this poorly understood bone dysplasia-mental retardation syndrome.
Acknowledgments
The first three authors contributed equally to this work. We are grateful to Stanislas Lyonnet for helping discussion and comments.
Electronic database information: Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/. National Center for Biotechnology Information, http: //www.ncbi.nlm.nih.gov/. Human Genome working Draft, http://genome.ucsc.edu/.