Article Text

Abstract
Mutations of the PTEN gene are associated with hamartoma-neoplasia syndromes. While germline mutations at this chromosome 10q22-23 locus have been observed in patients with Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome (BRR), both of which phenotypes are associated with hamartomata and neoplasia, somatic mutation of PTEN has been established in a wide variety of sporadically occurring neoplasia. CS and BRR share some clinical features, specifically hamartomata and lipomatosis. Investigation of other clinically distinct syndromes associated with lipomatosis and overgrowth has established germline and germline mosaic PTEN mutations in several patients with Proteus syndrome. To this expanding array of clinically distinct phenotypes associated with PTENmutations, we now report a novel heterozygous germline mutation, H61D, in a patient with features of VATER association with macrocephaly and ventriculomegaly.
- VATER
- hydrocephalus
- PTEN
Statistics from Altmetric.com
The PTEN locus on chromosome 10q22-23 encodes an almost ubiquitously expressed dual specificity phosphatase which acts as a tumour suppressor.1-3Mutations at this locus have been established as the cause of Cowden syndrome (CS), an autosomal dominant disorder characterised by macrocephaly and age related emergence of hamartomas and which also bears a high risk of breast and thyroid cancers.4 5Similarly, mutations of PTEN have been established in Bannayan-Riley-Ruvalcaba syndrome (BRR), an autosomal dominant developmental disorder characterised by macrocephaly, developmental delay, lipomatosis, and haemangiomatosis, and which characteristically has spots on the prepuce of the penis in affected males.6 7 Arising from these observations, there has been a heightened awareness of the variability in phenotypic presentation of patients at risk of developing the breast and thyroid malignancy characteristically associated with CS and revised diagnostic criteria have been proposed in addition to recommendations for optimal surveillance.8
Further phenotypic variation of PTEN has been heralded by the identification of a germline R335X mutation in a patient with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations, and lipomatosis.9A second mutation, R130X, was identified in the opposite allele in several abnormal tissues comprising a lipomatous mass, an epidermoid naevus, and an arteriovenous malformation from distinct sites in the same patient. Both mutations are known to be pathogenic, being described in CS and BRR. These findings suggesting a causative role forPTEN mutation in patients with Proteus syndrome and overlapping clinical syndromes of overgrowth have been substantiated in a recent study.10
In similar vein, several reports have drawn attention to patients with an underlying diagnosis of Fanconi anaemia but whose clinical presentation was of VATER association with hydrocephalus.11-14 Similarly, reports of affected sibs with the VATER-hydrocephalus phenotype15 16 and parental consanguinity in the parents of three affected males17support a likely autosomal recessive single gene aetiology in at least a proportion of cases with this phenotype. Moreover, increased sister chromatid exchange and increased chromosome breakage in response to alkylating agents have been reported in this phenotype, further substantiating a possible link with Fanconi anaemia and, indeed, mutation of the FAC gene has been observed in affected twins.11 14 18 However, the complex aetiology of the VATER-hydrocephalus phenotype is signalled by reports consistent with X linked inheritance18 and by variations in the phenotype reported, which have included abnormal ears, branchial arch defects, aqueduct stenosis, pancreatic hypoplasia, and abnormal lung lobulation.16 17 19 Savarirayanet al,20 in describing a case of Baller-Gerold syndrome, emphasise the potential for diagnostic error in making a diagnosis of Baller-Gerold syndrome or VACTERL association in children with this spectrum of anomalies without first excluding conditions such as Fanconi anaemia and Roberts syndrome, for which diagnostic tests are available. As recognised by Winter and Baraitser,19 “it seems very likely that there is genetic heterogeneity amongst the group of infants with hydrocephaly and features of VATER”. The observation we now report, of a novelPTEN missense mutation, H61D, in a patient with features of VATER association in whom macrocephaly and ventriculomegaly are also documented further underlines the likely genetic heterogeneity in patients with this phenotype.
Methods and results
PHENOTYPIC DESCRIPTION
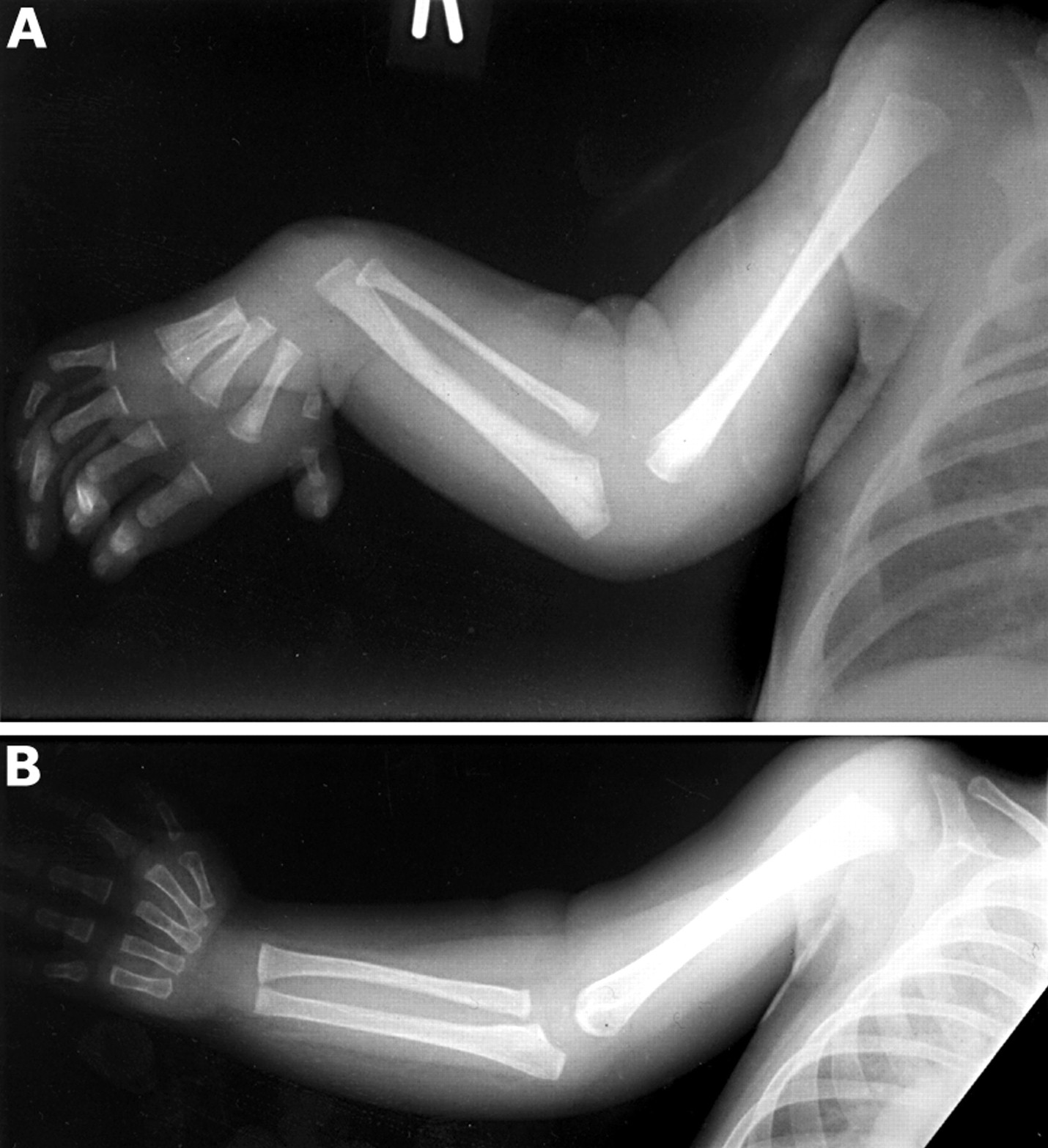
A male patient, the second offspring of his non-consanguineous parents, was identified during gestation as probably having a tracheo-oesophageal fistula, owing to polyhydramnios and an absent stomach bubble on ultrasonography. Delivered at 37 weeks, birth weight was 2940 g (50th centile). Macrocephaly was present at birth, OFC being 37.5 cm, the 97th centile being 35 cm. Echocardiography showed a normal heart. Bilateral hand malformations were noted neonatally, specifically hypoplasia of the thumbs bilaterally with radial deviation of the hands, more marked on the right side (fig 1). Clinically, the macrocephaly has persisted. Aged 18 months, OFC is 55 cm, almost 5 cm above the 97th centile. There are no skin pigmentary signs. There are no cutaneous stigmata of CS or BRR. The external ears and anus are unremarkable.

Clinical observation of the hands shows proximally inserted hypoplastic thumb on the right with absence of the thenar eminence (A). On the left, the thenar muscle is also reduced and the thumb hypoplastic and immobile at the interphalangeal joint with absence of the skin crease (B).
INVESTIGATIONS
Radiological investigation of this patient has shown normal renal, ureteric, and bladder anatomy. There are no vertebral malformations but 13 pairs of ribs are seen. The left upper limb shows slightly small first metacarpal and phalanges of the thumb (fig 2), while on the right side the radius is hypoplastic as are the first metacarpal and the phalanges of the thumb (fig 2). Initial CT scan, aged 2 weeks, showed no evidence of ventriculomegaly, but subsequent re-evaluation by CT scan aged 3 months showed that a fullness of both lateral ventricles had developed. Subsequent monitoring has confirmed a non-progressive ventriculomegaly.
The right upper limb (A) showing a gracile radius and markedly reduced first metacarpal and phalanges of the thumb. Note the normal forearm bones and slight shortening of the first metacarpal and phalanges of the thumb of the left upper limb (B).
There is no evidence of craniosynostosis onx ray. Routine karyotype is normal. There is no heterochromatin puffing. There is no evidence of enhanced chromosome fragility on exposure to alkylating agents.
PTEN ANALYSIS
Mutation analysis of the PTEN locus has shown a de novo germline mutation, H61D (fig 3), not present in either parent and paternity is confirmed.

{kind=link}
{kind=link}
{kind=link}
Sequencing chromatogram showing the wild type (WT) and heterozygous mutant (H61D) sequence at codon 61 of PTEN.
Discussion
VATER association is the term given to the non-random development of vertebral, anal, radial, and renal malformations in patients with TOF or oesophageal atresia.21 The wide spectrum of associated malformations can extend beyond those structures indicated in the acronym, pulmonary agenesis being a good example.22-24 Patients with features of VATER association but who, in addition, have hydrocephalus do appear to represent a distinct group genetically and phenotypically. Firstly, the familiality of several reports of VATER with hydrocephalus stands in contrast to the sporadic occurrence of the VATER association devoid of hydrocephalus.11-17 Secondly, the observation of cytogenetic characteristics consistent with Fanconi anaemia in several subjects with the VATER-hydrocephalus presentation11-13and the identification of an FAC gene mutation in one such family14 is quite distinct from uncomplicated VATER association, in which cytogenetic studies are generally unremarkable. Finally, the scope for diagnostic confusion offered by the VATER-hydrocephalus phenotype, in particular with Baller-Gerold syndrome, Fanconi anaemia, and Roberts syndrome, has been underlined by the experience of several authors,11 12 15 18 20 25 26 some families having gone through the full spectrum of these diagnostic entities before the true diagnosis became apparent.13
Against this backdrop, we now report a novel PTENgermline mutation in a patient with macrocephaly, ventriculomegaly, TOF, and bilateral radial hand anomalies. While the subject of this report does not represent a classical example of VATER association, he does significantly resemble those patients described with the VATER-hydrocephalus phenotype. Previous observations in patients with PTEN mutations have established that macrocephaly is a frequent feature rather than an exception among that cohort.8 However, nothing in previously published reports of PTENmutation has suggested that alteration at this locus might offer an explanation for TOF and radial malformations in patients with a VATER-ventriculomegaly phenotype. Accordingly, we have had to consider the possibility that our observation of PTENmutation in this patient is a chance finding of no aetiological significance. However, several lines of evidence argue against such an interpretation.
Asymmetrical limb malformations have been seen in those cases of Proteus syndrome and clinically overlapping malformation syndromes in which a range of mutations at the PTEN locus have been described.9 10 Given the ubiquitous expression pattern of PTEN in human tissue3 and during embryonic and fetal development27 allied to these limb malformations in the patients described with Proteus syndrome andPTEN mutation,9 10 it seems plausible that there may be other phenotypes ofPTEN mutation associated with limb malformation. Missense mutations comprise 20% of thePTEN mutational spectrum among CS and BRR patients5 and are also described in association with Proteus and clinically overlapping syndromes.9 10 There are approximately 22 different missense mutations inPTEN reported to date.10 28 So far, every missense mutation is believed to be pathogenic and at least eight have been shown to be phosphatase dead (complete loss of wild type phosphatase activity).29 Of these 22 missense mutations, 16 lie within the first five exons. Those within the phosphatase core motif disrupt phosphatase function probably by a dominant negative mechanism.30 The particular missense mutation which we describe, arising as a new genetic event in this report, has not been previously described. However, there are strong grounds to suggest that this mutation is pathogenic. Firstly, the mutation is in exon 3, which already harbours four other missense mutations, three of which are in close proximity to this mutation (to date, there are no germline truncating mutations in exon 3).5 28 Secondly, the H to D substitution, representing the replacement of a heterocyclic basic hydrophilic amino acid by an aliphatic, polar, acidic dicarboxylic amino acid, is a non-conservative change. Indeed, a different missense mutation at codon 61, H61R, has been shown to be phosphatase dead in functional assays.30Thirdly, exon 3 encodes part of the tensin and auxilin homology domain. Thus, disruption of this highly conserved domain would probably cause disruption of cell adhesion and cell motility. Therefore, we would suggest that this germline PTEN mutation is highly likely to be pathogenic and has caused the clinical features in this patient, including the VATER-hydrocephalus association.
The VATER-hydrocephalus phenotype offers particular diagnostic and counselling challenges to the clinical geneticist and, as exemplified by the experiences of several authors, can be confusing. Based on our experience in this case, analysis of PTENmay prove to be instructive in a larger series of such cases and may ultimately prove useful in identifying the aetiology in a proportion of patients with this heterogeneous phenotype.