Article Text

Abstract
The main features of Silver-Russell syndrome (SRS) are pre- and postnatal growth restriction and a characteristic small, triangular face. SRS is also accompanied by other dysmorphic features including fifth finger clinodactyly and skeletal asymmetry. The disorder is clinically and genetically heterogeneous, and various modes of inheritance and abnormalities involving chromosomes 7, 8, 15, 17, and 18 have been associated with SRS and SRS-like cases. However, only chromosomes 7 and 17 have been consistently implicated in patients with a strict clinical diagnosis of SRS. Two cases of balanced translocations with breakpoints in 17q23.3-q25 and two cases with a hemizygous deletion of the chorionic somatomammatropin gene (CSH1) on 17q24.1 have been associated with SRS, strongly implicating this region. Maternal uniparental disomy for chromosome 7 (mUPD(7)) occurs in up to 10% of SRS patients, with disruption of genomic imprinting underlying the disease status in these cases. Recently, two SRS patients with a maternal duplication of 7p11.2-p13, and a single proband with segmental mUPD for the region 7q31-qter, were described. These key patients define two separate candidate regions for SRS on both the p and q arms of chromosome 7. Both the 7p11.2-p13 and 7q31-qter regions are subject to genomic imprinting and the homologous regions in the mouse are associated with imprinted growth phenotypes. This review provides an overview of the genetics of SRS, and focuses on the newly defined candidate regions on chromosome 7. The analyses of imprinted candidate genes within 7p11.2-p13 and 7q31-qter, and gene candidates on distal 17q, are discussed.
- Silver-Russell syndrome
- imprinting
- mUPD(7)
- candidates
Statistics from Altmetric.com
Silver-Russell syndrome (SRS) is a clinically and genetically heterogeneous disorder of growth with a spectrum of additional dysmorphic features. SRS may comprise different disorders with clinically similar phenotypes or may result from disruption of different components of a single biochemical or endocrinological pathway, in either case reflecting its genetic heterogeneity. This review focuses on the influence of genomic imprinting in SRS and recent progress in defining two candidate SRS regions on both the p and q arms of chromosome 7. The investigation of imprinted genes on chromosome 7 and candidate genes from other chromosomal locations are also discussed with regard to their potential role in SRS.
Clinical features of Silver-Russell syndrome
SRS is a congenital disorder characterised by intrauterine and postnatal growth retardation in association with a number of dysmorphic features first described in 1953 by Silver et al 1 and in 1954 by Russell.2 The syndrome is clinically heterogeneous and diagnosis may be subjective. Two recent reviews provide details of the spectrum and frequency of the various clinical manifestations of SRS in two large groups of patients.3 4 Wollmann et al 3 additionally reviewed the growth characteristics of 386 SRS patients. Table 1 lists the clinical features of SRS and their frequencies, as determined in these two reviews.3 4 Low birth weight, short stature resulting from postnatal growth retardation, and a characteristic, small, triangular face were observed in the vast majority of patients. Clinodactyly of the fifth finger, relative macrocephaly owing to sparing of cranial growth, and facial, limb, or body asymmetry were also frequently observed. Although no strict diagnostic criteria for SRS have been established, presence of the three major features plus one or more of the minor confirmatory features (such as clinodactyly or asymmetry) are generally required for a positive diagnosis.3-5 However, none of these traits is absolute and there are a small number of diagnosed SRS patients in whom a major feature is absent, but several of the minor ones are present. A recent novel observation is the presence of bilateral camptodactyly with terminal interphalangeal contractures in 10 of 31 classical SRS patients.4
Clinical features and their reported frequencies in SRS. and genetic subgroups of SRS. The percentage of SRS features in two separate groups of SRS patients are listed.3 4 These figures could not be combined as certain clinical conditions were not examined consistently in both sets of patients. These serve as a reference for the frequency of features observed in specific genetic categories of SRS patients mentioned in the text. Where the patient number is small the frequencies are given as fractions, not percentages. Traits either not reported or not evaluated are blank. Non-SRS features apparent in the SRS-like patients with abnormalities of chromosomes 18 and 8 are not included, but discussed in the text
Genetic heterogeneity
The majority of SRS cases are sporadic.5 However, in a review of 197 SRS probands, 19% had more than one affected person in the family, providing evidence for a genetic cause.6Different modes of inheritance have also been suggested on the basis of family histories, indicating that SRS is genetically heterogeneous (reviewed by Duncan et al 6). Autosomal recessive transmission appears likely in several families in which there is more than one affected sib but the parents are phenotypically normal. Consanguinity between the normal parents in four unrelated Arab families with two or more SRS offspring supports transmission of an autosomal recessive mutation in these cases.7 8 Autosomal and X linked dominant inheritance patterns have also been suggested in SRS.6 9 10 In their review, Duncan et al 6 describe 23 families with 38 SRS patients. In 17 of these, multiple maternal relatives either had full SRS or a partial phenotype.6 Autosomal dominant inheritance, with intrafamilial variation in expression owing to incomplete penetrance or gene pleiotropy is one possibility. Alternatively, the primarily female transmission of this disorder in these families is suggestive of an X linked dominant form. While paternal inheritance of SRS has been described, no male to male transmission has been documented.6 10 Hypogonadism accompanying SRS in males may be a factor in reducing male transmission of SRS. Nevertheless, this means that X linked dominant and autosomal dominant inheritance patterns may be indistinguishable in these families.6 In view of recent evidence to suggest that genomic imprinting plays a role in certain SRS patients,11an imprinted mode of inheritance may be invoked in some SRS families with more than one affected. Certainly the predominant maternal transmission of SRS is compatible with a matrilineal mode of inheritance owing to genomic imprinting.
Concordance for SRS in a pair of monozygotic twins and discordance in dizygotic twins lends some weight to a genetic basis for this syndrome (reviewed by Duncan et al 6). However, the report of a pair of monozygotic twins discordant for SRS is confounding. This may be solely because of environmental factors. Placental pathology, including hypotrophy, a single umbilical artery, and velamentous cord insertion was observed in the growth restricted twin fetus during pregnancy.12 However, the continuation of growth retardation postnatally is difficult to explain if purely environmental circumstances are involved. Alternatively, genetic factors such as a postzygotic mutation event or mosaicism, in concert with environmental factors, may be the cause of the disease phenotype in only one twin.
While genetic factors are evident in familial cases of SRS, the genetic contribution to the majority of isolated patients is difficult to assess. It is possible that some of these patients who are karyotypically normal have inherited recessive mutations, or have new dominant mutations, although there is no evidence of a parental age effect to support the latter.5
Structural chromosome abnormalities in patients with SRS or SRS-like features
The majority of SRS patients have a normal karyoptype. However, a small number of patients with SRS, or features reminiscent of SRS, have been described with structural abnormalities of different chromosomes, further illustrating the genetic heterogeneity of this disorder. These chromosomal disruptions provide clues regarding the location of the genes involved in SRS, aiding a positional cloning or positional candidate approach to identifying these. Several candidate genes mapping in these regions have been identified and analysed for involvement in SRS.
CHROMOSOMES 18 AND 8 WITH SRS-LIKE PHENOTYPE
A single case with SRS has been reported in association with trisomy 18 mosaicism.13 The diagnosis was made on the basis that the patient had the three major SRS features plus asymmetry (table 1), but additionally presented with characteristics of the trisomy 18 syndrome including low set ears, developmental delay, failure to thrive, a prominent occiput, and characteristic dermatoglyphics. The genital anomalies observed in this patient are common to both disorders.13 Several cases of trisomy 18 mosaicism have been documented with a SRS-like phenotype including low birth weight and congenital asymmetry, but these may represent overlapping features between the SRS and trisomy 18 syndromes.14-16
One patient diagnosed with SRS on the basis of IUGR and several minor SRS features (listed in table 1) has been described with a deletion of the short arm of chromosome 18. However, this patient did not have short stature. Furthermore, the facial dysmorphism, large, low set ears, and mental retardation observed were atypical of SRS.17 This patient was lacking two major SRS features so does not strictly fit the SRS phenotype.
One SRS-like patient has been described with a paternally inherited deletion of chromosome 8q11-13. This patient had all the major features of SRS and fifth finger clinodactyly, but was microcephalic and had additional non-SRS features.18 It is likely that the SRS characteristics observed in cases with chromosome 8 and 18 anomalies are a subset of the multitude of clinical manifestations displayed. These patients are likely to have a distinct non-SRS clinical entity, which merely shares some of the SRS features. Thus, it is debatable as to whether these cases should still be considered SRS, as they tend to cause confusion rather than clarity when considering genetic aetiology.
RING CHROMOSOME 15 AND DELETIONS OF 15q
Two patients with a distinct SRS phenotype have been identified with a r(15).19 20 Both patients had the three major SRS features and additional minor ones (table 1). A hemizygous deletion of the insulin-like growth factor receptor I gene (IGFIR), localised to 15q26.3, was identified in one of these cases analysed molecularly.20
A number of SRS-like patients have also been described with deletions of distal 15q or a ring chromosome 15 (r(15)) with accompanying deletions of 15q subbands.21-24 Several SRS features overlap with those seen in patients with the r(15) syndrome including IUGR, short stature, triangular facies, digital anomalies such as fifth finger clinodactyly, and café au lait naevi. However, there are differences between the two syndromes. Patients with a r(15) may additionally present with microcephaly, hypertelorism, and mental retardation not common to SRS, with absence of other SRS characteristics such as skeletal asymmetry, downturned corners of the mouth, and precocious puberty.21 23 24
The size of the deleted sections of distal 15q in r(15) syndrome patients are variable, but where these includedIGFIR, the patients were severely pre- and postnatally growth retarded.21 23 24 The insulin-like growth factor (IGF) family, consisting of insulin, IGF1 and IGF2, their receptors IGF1R and IGF2R, and six IGF binding proteins (IGFBPs), plays a crucial role in regulation of fetal and neonatal growth and development.25 26 It was hypothesised that hemizygous loss of the IGFIR gene was responsible for the phenotype in the two SRS patients with r(15) and the shared features between SRS and r(15) syndrome patients.IGFIR was also proposed as a candidate for SRS in patients with normal karyotypes.20 However, no hemizygous deletions were identified in a total of 38 such SRS patients, screened in two separate studies.24 27Furthermore, no mutations were identified in two exons encoding crucial active sites within the protein in 33 probands, indicating that this gene cannot be largely responsible for SRS in other groups of patients.27
DISTAL CHROMOSOME 17q
Two patients with severe SRS have been described with reciprocal translocations involving distal chromosome 17q, with the breakpoints originally localised to 17q25.28 29 Both patients had several minor SRS traits in addition to the three major criteria (table1). The first case had an apparently balanced translocation (17;20)(q25;q13) inherited from the phenotypically normal father.28 The second case had a de novo translocation (1;17)(q31;q25).29 The translocation breakpoint in the latter case has now been cloned and the localisation refined to 17q23.3-q24.30
Two members of the growth factor receptor bound protein (GRB) family of genes, GRB2 andGRB7, which map to 17q25.1 and 17q21.1 respectively, have been analysed as candidates for SRS. The GRB proteins interact with various receptor tyrosine kinases within the insulin and IGFI signal transduction axes, and so play a role in mitogenesis (reviewed by Daly et al 31). GRB2 and the guanine nucleotide exchange factor, Sos, form a complex, which interacts with the insulin receptor substrate I (IRS-I), and thus regulates Ras activation in the first steps of the insulin signalling pathway.32 Minor structural anomalies of GRB2 andGRB7 have been excluded in 36 SRS patients with normal karyotypes (M Hitchins, unpublished data).GRB2 was screened for mutations of the coding region by two groups, in a total of 29 SRS patients, but none was found, indicating that these genes do not play a significant role in SRS33 (M Hitchins, unpublished data). However, the potential effects of the two reported translocations on expression of these genes has not been investigated. The growth hormone (GH) gene cluster, including the GH gene and the chorionic somatomammotropin hormone (CSH) genesCSH1 and CSH2, are located on chromosome 17q24.1.34 A patient with classical SRS (table 1) has been reported with a paternally inherited deletion of the CSH1 gene.35Recently, a second case with a similar deletion has been identified (T Eggermann, personal communication). CSH1, or placental lactogen, is produced in the placental syncytiotrophoblast and secreted into the fetal and maternal circulation. Low levels of placental lactogen during pregnancy have been associated with pathological conditions including IUGR, diabetes, and pre-eclampsia, but these may be because of associated factors such as placental infarction restricting maternal-fetal circulation (reviewed by Handwerger36). Several cases with normal phenotypes at birth have been reported with absence of placental lactogen during pregnancy owing toCSH1 deletions on both chromosome 17 homologues in the fetus.37 38 Thus, the significance of the hemizygous CSH1 deletion in the two SRS patients is unclear, but may have contributed to the syndrome in combination with other genetic or environmental factors. Infarction of the placenta, for instance, was noted in the first case.35Deletion of CSH1 in association with SRS is rare and similar deletions have been ruled out in 106 SRS patients to date35 (T Eggermann, personal communication; M Hitchins, unpublished data).
Chromosome 7 and genomic imprinting in SRS
UNIPARENTAL DISOMY AND GENOMIC IMPRINTING
Uniparental disomy (UPD) is the rare inheritance of both copies of one particular chromosome from a single parent.39 UPD occurs through two main mechanisms, following a non-disjunction event during meiosis. (1) Fusion of a disomic gamete with a normal monosomic one, with subsequent loss of one of the extra homologues from the trisomic conceptus, is termed “trisomic rescue”. Uniparental heterodisomy, which is the inheritance of two different homologues from one parent, that is, both grandparental copies, arises through this mechanism. This may also be accompanied by the observation of trisomy mosaicism in the placenta.40 (2) Fusion of a nullisomic gamete with a normal one forming a monosomic zygote, followed by mitotic duplication of the existing homologue in the conceptus, is termed “monosomy rescue”. This mechanism results in two replica copies of the original parental homologue, referred to as uniparental isodisomy. UPD may be a mixture of hetero- and isodisomy owing to recombination at meiosis I. UPD can also occur through a mitotic error after fertilisation. This may result in segmental disomy following somatic recombination, or may be accompanied by trisomy mosaicism or partial supernumerary chromosomes.41-43
Genetic disorders as a consequence of UPD may occur through two distinct mechanisms. (1) Recessive mutations can be reduced to homozygosity owing to isodisomy of the locus if the transmitting parent is a heterozygous carrier. (2) Disruption of expression of genes subject to genomic imprinting can occur, in both iso- and heterodisomy, where the chromosome in question contains an imprinted region.
Genomic imprinting is defined as the differential expression of a gene or chromosomal region according to the parental origin of inheritance. Imprinted genes are expressed from a single parental allele (fig 1A), such that in UPD there may be absence of an active copy on the parental homologue that is missing, or overexpression on the duplicate parental chromosome (fig 1B). Imprinted genes tend to be clustered together in regions in which their expression is co-ordinately regulated and are associated with sites of parental allele specific methylation of the DNA. A regularly updated catalogue of imprinted genes may be found atwww.otago.ac.nz/IGC.44 Imprinted genes and regions are also generally conserved in evolution. Imprinted regions in the mouse have been defined by the use of reciprocal translocations to derive embryos that are uniparental for specific subchromosomal regions, which give rise to a noticeable phenotype.45 There are currently 14 defined regions of “non-complementation”, which require both parental complements for normal growth and development, spread over eight different mouse chromosomes. A map of these imprinted regions and genes in the mouse, with the human homology map, is available atwww.mgu.har.mrc.ac.uk/imprinting/imprinting.html.
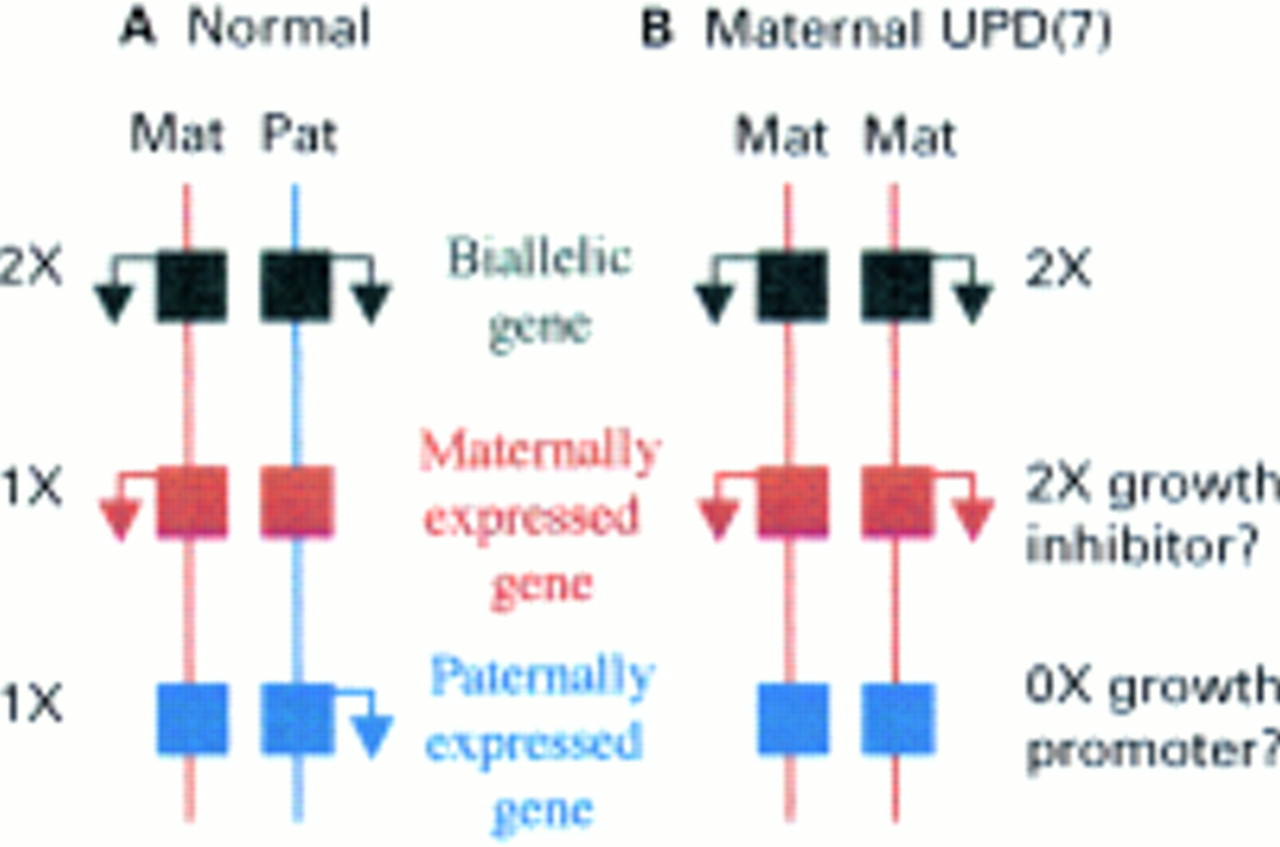
Possible consequences of maternal uniparental disomy for chromosome 7 in Silver-Russell syndrome. Maternal and paternal chromosome homologues are depicted by vertical lines in red and blue, respectively. Genes are represented by squares and arrows denote transcriptional activity. Expression of biallelic genes are not affected by mUPD. There is a double dose of maternally expressed imprinted genes in mUPD. If one of these is involved in growth suppression it might account for SRS. There is absence of transcription of paternally expressed imprinted genes in mUPD as these are silent on the maternal allele. Absence of a paternally expressed growth promoter could cause SRS.
MATERNAL UNIPARENTAL DISOMY FOR CHROMOSOME 7 AND SRS
Maternal UPD for chromosome 7 (mUPD(7)), that is, the inheritance of both homologues from the mother, occurs in 7-10% of SRS cases.46 47 The first cases of mUPD(7) were ascertained through diagnosis of recessive disorders, but prompted researchers to screen for UPD(7) among the SRS population. Two cases with cystic fibrosis (CF) resulting from maternal isodisomy for chromosome 7, for which the mothers were CFTR mutation carriers, were the first cases of UPD to be described.48 49 Both patients had primordial growth retardation, which could not be attributed to CF alone, and other features of SRS. A third patient with a collagen disorder and marked short stature was found to have mixed maternal hetero- and isodisomy for chromosome 7, with a homozygous mutation of theCOL1A2 gene.50 Screens of entire SRS patient cohorts for mUPD(7) have subsequently been carried out and at least 40 cases of mUPD(7) in association with SRS (or SRS features) have been published.40 43 46-56 Some researchers excluded mUPD(7) in their group of patients,57which helped in determining the frequency of this occurrence more accurately. UPD involving chromosomes 2, 6, 9, 14, 16, 20, and 22 in association with SRS have also been investigated, but ruled out.55 57 58
Some cases of SRS with mUPD(7) have a mild or incomplete phenotype.4 47 59 Features appearing milder or less frequently in mUPD(7) patients include the facial dysmorphism, asymmetry, clinodactyly and other digital abnormalities, café au lait spots, precocious puberty, squeaky voice, and tooth anomalies, as listed in table 1.51 52 59 However, mUPD(7) patients experienced severe feeding difficulties in the first few years of life, speech delay, and excessive sweating. These patients may comprise a more homogeneous group that are clinically distinguishable from other SRS patients.4 47 59
MECHANISM CAUSING SRS IN mUPD(7)
The mechanism by which mUPD(7) causes SRS has been investigated. Cases with biparental inheritance of chromosome 7, but with confined placental mosaicism for trisomy 7 as a consequence of “trisomic rescue” were found to have normal birth weights. This excluded the possibility that IUGR in SRS is the result of malfunction of a trisomy 7 placenta.40 Two patients with paternal UPD(7) (pUPD(7)) have been reported with recessive disorders owing to isodisomy at mutant recessive loci. The first patient had congenital chloride diarrhoea but had normal growth patterns.60 The second child had CF with complete situs inversus and immotile cilia in addition, and had poor growth at 6 months of age most likely as a consequence of his recessive phenotype.61 The lack of a distinct growth phenotype in the patients with pUPD(7), in contrast to the consistent severe growth retardation seen in mUPD(7), implicates imprinted genes in the aetiology of SRS in these cases.
To differentiate between unmasking of a recessive allele by isodisomy and disruption of genomic imprinting as the cause of SRS, Preeceet al 11 analysed the full length of the chromosome 7 homologues in five mUPD(7) cases for regions of consistent isodisomy.11 Forty polymorphic markers distributed along the entire chromosome 7, at an average genetic interval of 4.5 cM, were analysed in each patient. The chromosomes 7 were mixed hetero- and isodisomy in each case, but no region of isodisomy was common to all five patients. This study excluded recessive mutations as the cause of SRS in cases with mUPD(7), indicating that an imprinting defect is the most likely cause for this disorder.11 SRS could be caused by the absence of expression of a gene involved in growth promotion that is active exclusively on the paternal allele, or by excess of a maternally expressed imprinted gene that is involved in growth inhibition (fig1B).
Two separate candidate regions for SRS on chromosome 7
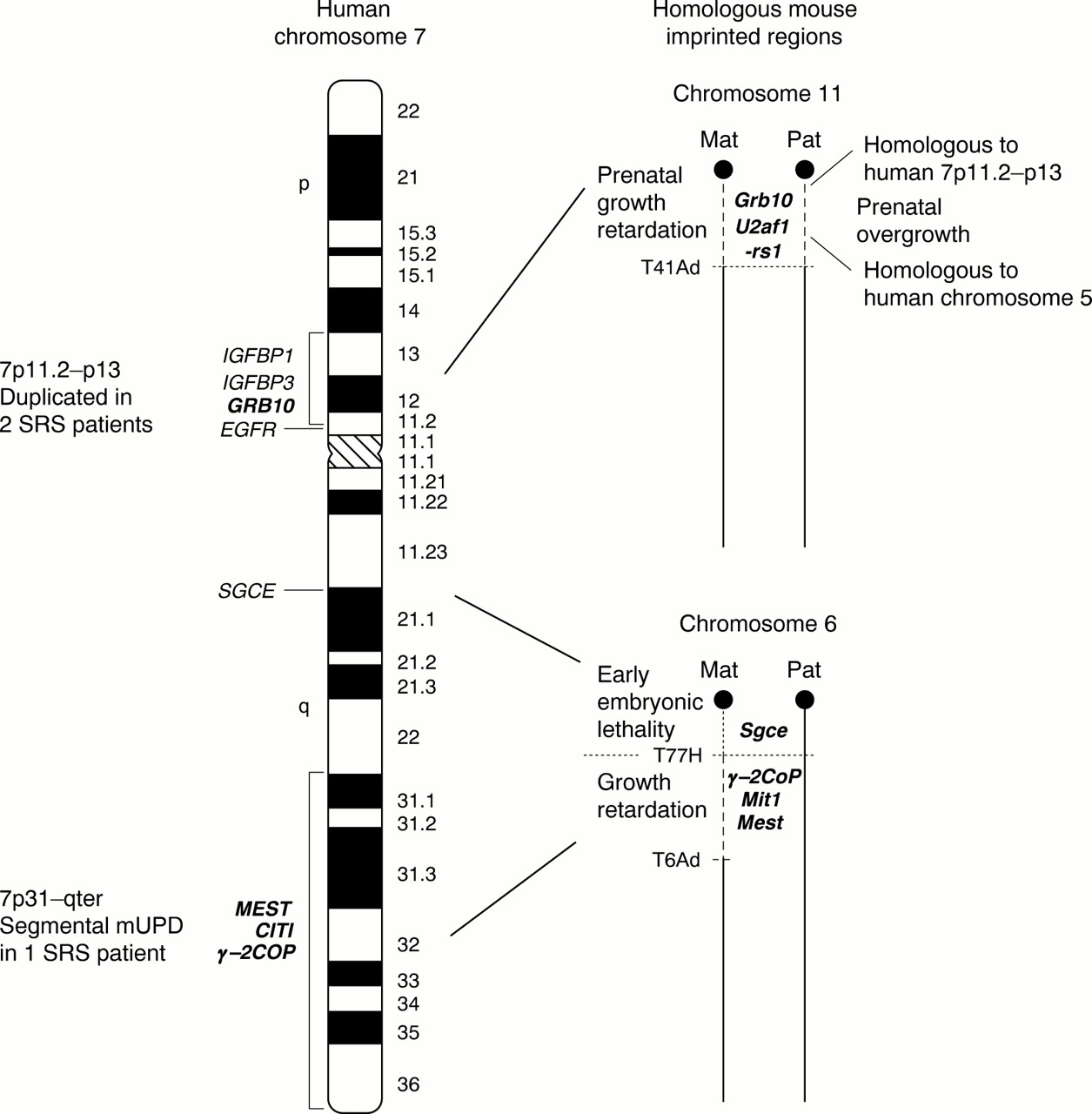
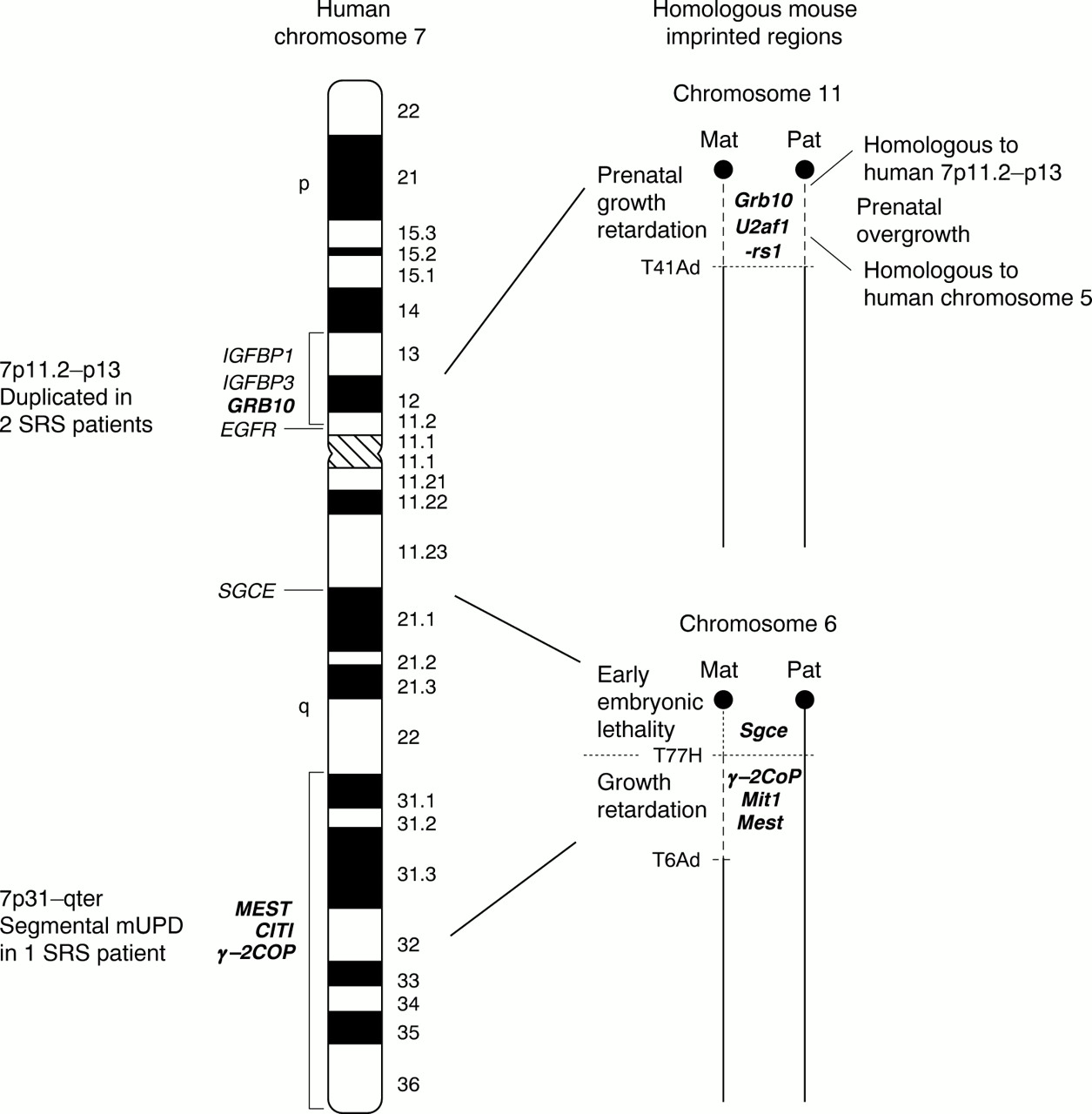
Maternal UPD(7) in SRS has involved the whole chromosome in the majority of cases. Thus, until recently, regions of human chromosome 7 that were homologous to imprinted regions in the mouse provided the main clue as to the location of imprinted genes that play a role in SRS. Different segments of human chromosome 7 share homology with regions from 10 different mouse chromosomes including 2, 4, 5, 6, 7, 10, 11, 12, 13, and 16. These are detailed atwww.ncbi.nlm.nih.gov/Homology/human7.html/. However, just two regions on human 7 share homology with definitive mouse imprinted regions; human 7p11.2-p13 is homologous to mouse proximal chromosome 11 and human 7q21-qter has a conserved region on proximal mouse chromosome 6 (fig 2). Both these regions in mice are associated with growth phenotypes. These regions aid in the selection of candidate genes for SRS and enable the study of imprinting processes that may contribute to SRS in the mouse system.
{kind=link}
{kind=link}
Candidate imprinted regions for Silver-Russell syndrome on human chromosome 7 and homologous imprinted regions in mouse. An ideogram of human chromosome 7 is shown with vertical black bars indicating the two SRS candidate regions on both the p and q arm. Candidate (or previous candidate) genes for SRS are labelled on the left in their approximate mapping locations. Imprinted genes are in bold. Mouse homologous regions to the two SRS candidate regions are depicted on the right. Mouse chromosome regions displaying non-complementation phenotypes with two maternal (and no paternal) copies and two paternal (and no maternal) copies are shown by dashed lines. These regions are defined by the mouse reciprocal translocations T(11;13)41Ad (chromosome 11),64 T(4;6)77H, and T(6;13)6Ad (chromosome 6).82 The approximate positions of mouse imprinted gene homologues are also indicated.
Recently the identification of key SRS patients with anomalies of 7p11.2-p13 and 7q31-qter has defined these two intervals as separate candidate regions for SRS. It is interesting to note that these two segments share homology with the aforementioned mouse imprinted regions.
THE 7p11.2-p13 REGION AND CANDIDATE GENES
Two patients with SRS have been identified with duplications of 7p12.1-p13.62 63 In the first case, the proband and her mother had an inverted interstitial duplication of this region. The SRS proband had relatively low birth weight, marked short stature, mild facial asymmetry, and fifth finger clinodactyly. The mother, whose duplication had arisen de novo on the grandpaternal chromosome, also had mild features of SRS, including short stature and fifth finger clinodactyly.62 The second report was of a de novo tandem duplication of the region 7p11.2-p13 on the maternally derived homologue, in a SRS patient presenting with pre- and postnatal growth restriction, small triangular face, and fifth finger clinodactyly, but no asymmetry.63 In both cases the duplicated region encompassed the insulin-like growth factor binding protein 1 (IGFBP1) and the growth factor receptor bound protein 10 gene (GRB10). The epidermal growth factor receptor gene (EGFR) lay outside the duplications (fig 2).62 63 Each of these genes had previously been proposed as candidates for SRS as they are important in the regulation of growth and development and their murine homologues map within the conserved imprinted region on proximal mouse chromosome 11.
Mice with maternal disomy/duplication for proximal 11 (MatDp.prox11) are 30% smaller than their normal littermates, possibly representing the growth restriction seen in SRS. Conversely, mice with paternal disomy for this region (PatDp.prox11) are 30% larger than normal.64 Although pUPD(7) is not associated with a growth phenotype in humans, there are other imprinted genes within proximal mouse 11 whose human homologues map to chromosomes other than 7, which may account for the overgrowth in PatDp.prox 11 mice. For example, the paternally expressed U2af1-rs1 gene with a human equivalent U2AF1-RS1 on chromosome 5 is a candidate for this excessive growth in these mice (fig2).64
EGFR was previously shown to be an unlikely candidate for SRS on the basis that it was not imprinted during human fetal development.65 That EGFRis not within the duplicated region in the two recently described SRS patients corroborates this (fig 2).62 63 IGFBP1 and IGFBP3have similarly been shown to be expressed from both parental alleles in human fetal tissues, so were deemed unlikely to contribute to SRS.66 67 Furthermore, in a screen of 49 SRS patients, no mutations of either IGFBP1 orIGFBP3 were identified.68
The GRB10 gene was considered a prime candidate for SRS on the basis of its function as a growth inhibitor and its imprinting status in the mouse.GRB10 is a member of a family of growth factor receptor bound protein genes, which includesGRB2 and GRB7 on chromosome 17, and has at least four protein isoforms. GRB10 interacts with several tyrosine receptor kinase growth factor receptors including the insulin receptor, IGF1R, EGFR, and the platelet derived growth factor receptor, causing downregulation of growth.69 70 71 Grb10 was identified in a systematic screen for maternally expressed imprinted genes by subtractive hybridisation of cDNAs derived from a normal mouse embryo with those from an androgenetic embryo (with a paternal genetic complement). Expression specifically from the maternal allele was confirmed in the mouse.72 Imprinted expression ofGRB10 in human fetal development was found to be both tissue and isoform specific. A novel isoform was identified in fetal skeletal muscle, which was maternally expressed.73 In contrast, GRB10was paternally expressed in fetal brain and spinal cord, but biallelic in all other major fetal organs and tissues.73 74 The role that GRB10 may play in SRS is difficult to discern on the basis of this imprinting profile. Yoshihashiet al 75 reported a maternally transmitted P95S amino acid substitution inGRB10 in two unrelated Japanese SRS patients,75 but this was not found in a further 161 SRS subjects.76 This substitution was subsequently shown to be a rare polymorphism in the Japanese population and unlikely to be causative of SRS.77 No mutations ofGRB10 have been identified in a total of 139 SRS patients screened for the entire coding region of the gene, indicating that it does not contribute significantly to SRS.73-76 Disruption of imprinted expression ofGRB10, through altered methylation for instance, has not been investigated and remains a possibility. Since imprinted genes tend to occur in clusters, it is likely that other imprinted genes exist in the 7p11.2-p13 candidate region, which may be responsible for SRS in cases with and without mUPD(7).
Different opinions regarding whether the SRS phenotype in patients with a duplication of 7p11.2-p13 is attributable to an imprinting effect have been put forward. Monk et al 63 stated that duplication of this region in SRS provided evidence for involvement of a maternally expressed growth suppressor gene, as opposed to a growth promoter gene active on the paternal allele, assuming these cases have a common aetiology with mUPD(7) patients. However, others have suggested that SRS may be instead the result of an extra copy of this region.43 62The 7p12.1-p13 duplication in the SRS proband described by Joyceet al 62 was of paternal origin in the proband's mother, who also had SRS characteristics. However, the mother's clinical phenotype was mild and may have been caused by duplication or disruption of non-imprinted contiguous genes, which may contribute to some of the minor features associated with SRS. A SRS patient with maternal isodisomy 7 and ring chromosome 7 (r(7)) mosaicism has been reported. The r(7) consisted of the 7p11-q11 region and was identified as paternal in origin. It is most likely that mitotic duplication of the maternal 7 homologue occurred in the zygote that was essentially monosomic, with just a partial paternal chromosome 7 to correct for chromosome number. Mosaicism for the r(7) probably reflected the instability of supernumerary partial chromosomes during cell replication. Although mUPD(7) was considered the underlying cause of SRS, the authors suggested that genes in the 7p11-q11 region were not involved in SRS since a paternal copy was present.43However, the behaviour of genes in ring chromosomes is not clear, and so a paternal copy in some cells may not compensate fully for loss of a complete paternal homologue.78 It is plausible that partial trisomy for chromosome 7 may be responsible for aspects of the SRS phenotype in this patient, in the same way that trisomy mosaicism for distal chromosome 11 may cause hemihypertrophy in Beckwith-Wiedemann syndrome patients with segmental pUPD for 11p15.5.41 42 No evidence for low level trisomy 7 mosaicism has been identified in four SRS patients with mUPD(7), though further mUPD(7) patients should be screened, especially those with asymmetry, before this can be ruled out as a contributory mechanism for SRS.79
THE 7q31-qter REGION AND CANDIDATE GENES
Two patients with SRS or some of the major SRS features have been reported with mUPD restricted to the q arm of chromosome 7.80 81 This suggests that the imprinted genes responsible for SRS in patients with mUPD(7) for the entire chromosome are located on the q arm and that the p arm may not involve an imprinting aetiology. A unique patient was reported with isochromosomes of 7p and 7q, showing homozygosity at all chromosome loci investigated. Paternal isodisomy was found for the 7p arm and the 7q arm was maternally isodisomic. The patient did not have IUGR, but became growth retarded postnatally, had mild limb asymmetry, slight fifth finger clinodactyly, and a triangular shaped face. The authors hypothesised that a maternal contribution to the p arm was necessary for normal fetal growth in utero, but that imprinted genes on the q arm were responsible for the additional SRS features noted in this patient.80 Recently, a patient with typical SRS including severe IUGR, short stature, relative macrocephaly, slightly triangular face with downturned corners of the mouth, mild fifth finger clinodactyly, abnormal spacing of the teeth and a squeaky voice, but no asymmetry, was described with segmental maternal isodisomy for the region 7q31-qter. This report shows that mUPD for this small interval on chromosome 7 causes a clinically definitive SRS phenotype, including several of the minor features.81 The finding of mUPD restricted to 7q31-qter in this SRS patient indicates that two separate regions on chromosome 7 can independently cause SRS, with the telomeric region of the q arm most certainly involving an imprinting defect (fig2).
The human 7q21-qter region is homologous to proximal mouse chromosome 6, which shows two distinct imprinted phenotypes in mice with maternal UPD. These are defined by two translocation breakpoints, with the proximal region between the centromere and the T77H breakpoint, and the distal region between the T77H and T6Ad translocation breakpoints (fig2). The distal imprinting segment is associated with pre- and postnatal growth restriction in mice with maternal disomy.82 This region contains a cluster of imprinted genes including the paternally expressed mesoderm specific transcript gene (Mest/Peg1)83and the nonclathrincoatprotein gene (γ-2Cop), which is maternally expressed in mouse, in contrast to its human homologue. Two untranslated paternally expressed transcripts Mit1 andγ-2Cop antisense, which overlapγ-2Cop, have also been reported in mouse.84 Mest was first identified in mice by subtractive hybridisation of cDNAs from a normal mouse embryo with those from a parthenogenetic embryo (with just a maternal genetic component) in a screen for paternally expressed genes.83 Mice with a paternally inherited mutation of theMest gene have IUGR and the female offspring themselves show abnormal maternal behaviour, with impaired placentophagia.85 The human homologueMEST has thus been proposed and investigated as a candidate for SRS.86
Human MEST87 andγ-2 COP 88 both map to 7q32 (fig 2).MEST is paternally expressed in embryos and all major fetal organs, but was found to be biallelic in adult lymphocytes.89 90 Two different isoforms ofMEST have since been identified in lymphocytes, one of which is biallelically expressed while the other is transcribed specifically from the paternal allele. Functional absence of MEST was shown in SRS patients with mUPD(7) by analysing this latter isoform in patient lymphoblasts.91 However, no mutations ofMEST have been identified in 49 SRS patients. MEST has a differentially methylated 5′ CpG island, thought to regulate expression, with hypermethylation of the inactive maternal allele.90Although patients with mUPD(7) only have the methylated maternal alleles at this site, no epigenetic alterations were identified in 35 non-mUPD(7) SRS patients, suggesting that their regulation ofMEST was normal. These data indicated thatMEST is unlikely to play a major role in SRS, although it may still contribute to the phenotype in mUPD(7) patients.86 The γ-2 COP gene is paternally expressed in all major fetal organs, with the exception of brain and liver, where biallelic expression was noted. γ-2 COP was screened for mutations in 42 SRS patients. A substitution was observed in one SRS patient, which was not identified in 98 normal subjects, but this was present on the inactive maternally derived allele, so was unrelated to the SRS phenotype.88 A non-translated paternally expressed transcript, CIT1, contained within an intron of γ-2 COP, has also recently been reported.92 Intensive physical and transcript mapping within 7q32 will help to identify other genes which may be subject to imprinting in this region.93
Is 7q21 a novel imprinted region?
A novel imprinted region may exist at 7q21. This is suggested by the homology between this region and the proximal imprinted region on mouse chromosome 6, between the centromere and T77H translocation breakpoint (fig 2). Maternal disomy for this region causes early embryonic lethality.82 This segment contains the paternally expressed ε-sarcoglycan gene (Sgce).94 The human homologueSGCE maps to 7q21, but its imprinting status is unknown.95 Furthermore a paternally expressed gene,PEG10, has recently been identified which maps to 7q21 near to SGCE. PEG10 appears to be derived from a retrotransposon.96 Although these genes may be located within a third imprinted region on chromosome 7, this lies proximal to the candidate imprinted region defined by segmental mUPD of 7q31-qter in SRS, and no chromosomal anomalies involving 7q21 have been associated with SRS to date.
Conclusions
The Silver-Russell syndrome is both clinically and genetically heterogeneous, with different modes of inheritance and chromosomal locations involved. This makes identification of the genes responsible difficult. Recent progress has been made on chromosome 7, defining two separate candidate regions, 7p11.2-p13 and 7q31-qter. A definitive role for genomic imprinting has been indicated in the 7q31-qter region, as a result of the observation of maternal uniparental disomy specifically for this interval in a single SRS patient. This case suggests that the 7q31-qter region alone may be responsible for SRS in patients with mUPD for the entire chromosome 7. Two imprinted genes within the disomic region, MEST and γ-2 COP, have been investigated as candidates for SRS, but there has been no direct evidence to show either is responsible for the phenotype. Currently there are conflicting opinions regarding the underlying basis for SRS in patients with structural anomalies of the 7p11.2-p13 region, with some arguing against a role for imprinting. However, this region contains one imprinted gene, which has been implicated in SRS, GRB10, though its potential involvement in the disorder remains uncertain. It may be the case that both the 7q31-qter and 7p11.2-p13 candidate regions cause SRS through disruption of imprinting. Since imprinted genes tend to be clustered in groups, there are likely to be other imprinted genes in both chromosome 7 candidate regions, which will require investigation for a role in SRS. The identification and analysis of additional SRS patients presenting with unique chromosomal abnormalities involving chromosome 7 will further define the candidate intervals. It is plausible that, as a variable phenotype, no single gene is responsible for all the features of SRS in any person. SRS may be a contiguous gene disorder, or where imprinting is involved, be the result of disruption of expression of several co-regulated imprinted genes within an imprinted region. Identification of the gene or genes causing SRS in one of the associated chromosomal locations (in particular on 7p, 7q, or chromosome 17) should aid considerably in the identification of the other genes involved on different chromosomes, by providing information on the growth and developmental pathway affected.
Acknowledgments
We would like to thank Colin Beechey of the MRC Mammalian Genetics Unit, Harwell for his help with fig 2. Megan Hitchins is funded as a postdoctoral fellow by the Dunhill Medical Trust.