Article Text

Abstract
Neurofibromatosis type 2 is an often devastating autosomal dominant disorder which, until relatively recently, was confused with its more common namesake neurofibromatosis type 1. Subjects who inherit a mutated allele of the NF2 gene inevitably develop schwannomas, affecting particularly the superior vestibular branch of the 8th cranial nerve, usually bilaterally. Meningiomas and other benign central nervous system tumours such as ependymomas are other common features. Much of the morbidity from these tumours results from their treatment. It is now possible to identify theNF2 mutation in most families, although about 20% of apparently sporadic cases are actually mosaic for their mutation. As a classical tumour suppressor, inactivation of theNF2 gene product, merlin/schwannomin, leads to the development of both NF2 associated and sporadic tumours. Merlin/schwannomin associates with proteins at the cell cytoskeleton near the plasma membrane and it inhibits cell proliferation, adhesion, and migration.
- NF2
- vestibular schwannomma
- meningioma
- mosaic
Statistics from Altmetric.com
History
The neurofibromatoses consist of at least two distinct dominantly inherited disorders, neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). For many years, these conditions were inextricably linked as part of von Recklinghausen disease. NF2 was first described in 1822 by the Scottish surgeon, Wishart.1NF1 was fully delineated in the late nineteenth century by von Recklinghausen.2 However, Cushing3 first bracketed them together in 1916 and his standing was such that, despite reports that the conditions were quite different,4 another half century elapsed before this was widely recognised. Indeed, publications on neurofibromatosis before 1985 are liberally sprinkled with NF2 cases being described as part of von Recklinghausen disease.5 The conditions were eventually recognised as separate entities with the localisation of the respective genes to chromosomes 17 and 22.6 7 This was followed by the US National Institutes of Health (NIH) consensus statement in 1987.8 Since 1987, it has generally been believed that NF2 is a genetically homogeneous condition, with no evidence of exclusion of classical NF2 (bilateral vestibular schwannomas (VS)) from theNF2 locus on 22q, in initial studies on small numbers of families.9 This has been confirmed in all reports of classical NF2 patients with bilateral VS since the simultaneous isolation of the gene by two groups in 1993.10 11 However, the possibility of another closely linked gene that affects the NF2 phenotype cannot be excluded.
Incidence and prevalence
In the UK, the only large population based estimate of birth incidence for NF2 showed that 1 in 33-40 000 people would be born with a mutation in the NF2 gene.12 A smaller study in southern Finland estimated a birth incidence of 1 in 87 000.13 The only other estimate of birth incidence (1 in 50 000) was made at the 1987 NIH Consensus Conference on the Neurofibromatoses, but a derivation for this figure was not provided.8 Initial estimates of prevalence suggested that as few as 1 per million were affected in the USA.14 The actual diagnostic prevalence from the UK population based study is 1 in 210 000.12 The annual incidence rate is 1 per 2 355 000, which represents about one new case per year for each Health Region in the UK,12 or 100 cases per year in the USA. The birth incidence is significantly higher than the diagnostic prevalence because many cases do not develop features of the condition until the third decade or later, and many other cases die before this time. With improvements in early diagnosis and tumour treatment, the prevalence is likely to rise, reflecting an increase from the 15 year survival from diagnosis for UK NF2 patients 10 or more years ago.15
Genetics
Heredity in NF2 was first reported in 1920 by Feiling and Ward,16 who described a three generation family with VS. The autosomal dominant transmission was confirmed in a large family reported by Gardner and Frazier.4 Subjects who inherit a pathogenic mutation in the NF2 gene will almost always develop symptoms by 60 years of age15; very occasionally, patients will have apparent non-penetrance.17 Although the transmission rate is 50% in the second generation and beyond, the risk of transmission in an apparently sporadic case of NF2 is less than 50% because of mosaicism.18 There were initial indications of a maternal gene effect, with earlier age at onset in people who had inherited the NF2 from their mother,12 14 but this effect is most likely the result of reduced genetic fitness among severely affected males.12 There is little evidence for anticipation.12
Clinical manifestations
The hallmark of NF2 is the development of bilateral VS (fig 1). The other main tumour features are schwannomas of the other cranial, spinal, and peripheral nerves, meningiomas, both intracranial (including optic nerve meningiomas) and intraspinal, and some low grade central nervous system (CNS) malignancies (ependymomas and gliomas). Four large clinical studies have now confirmed this clinical picture14 15 19 20 (table 1). Our diagnostic criteria for NF221 are shown in table 2. The original NIH criteria have been expanded to include patients with no family history who have multiple schwannomas/meningiomas, but who have not yet developed bilateral 8th nerve tumours. Patients may present with cranial meningiomas or a spinal tumour long before the appearance of a VS.15 22 23 Since 50% of cases represent new mutations,12 24 the criteria are more inclusive but are still extremely unlikely to include chance associations of isolated disease features (see under Genetics).

MRI scan showing bilateral enhancing masses (VS) in the cerebellopontine angle.
Clinical characteristics of NF2 patients in four studies
Diagnostic criteria for NF2 (these include the NIH criteria8 with additional criteria21)
PRESENTATION
The majority of patients with NF2 present with hearing loss, which is usually unilateral at onset. The hearing loss may be accompanied or preceded by tinnitus. VS may also cause features such as dizziness or imbalance as the first symptom. A significant proportion of cases (20-30%) present with an intracranial meningioma, spinal tumour, or cutaneous tumour.14 15 19 25 Indeed, the first sign of more severe multi-tumour disease in early childhood is often a non-8th nerve tumour. Adult presentation is thus quite different from paediatric presentation, in which VS accounts for as little as 15-30% of initial symptoms.22 There also appears to be a tendency to mononeuropathy, particularly affecting the facial nerve, causing a Bell's-like palsy, which does not fully recover, years before the detection of a VS. Some children present with a polio-like illness with wasting of muscle groups in a lower limb, which again does not fully recover. In adulthood, a more generalised polyneuropathy occurs in about 3-5% of patients, often associated with an “onion bulb” appearance on nerve biopsy.12 26 This can progress, leading to severe muscle wasting and even death.
Ophthalmic features are also prominent in NF2. Between 60-80% of patients have cataracts,15 27 28 which are usually presenile posterior subcapsular lenticular opacities that rarely require removal. However, cortical wedge opacities may be present from near birth. Optic nerve meningiomas can cause visual loss in the first years of life and extensive retinal hamartomas can also affect vision.23 29 Misdiagnosis of both of these abnormalities as retinoblastoma has led to the eye being removed in the first few years of life.
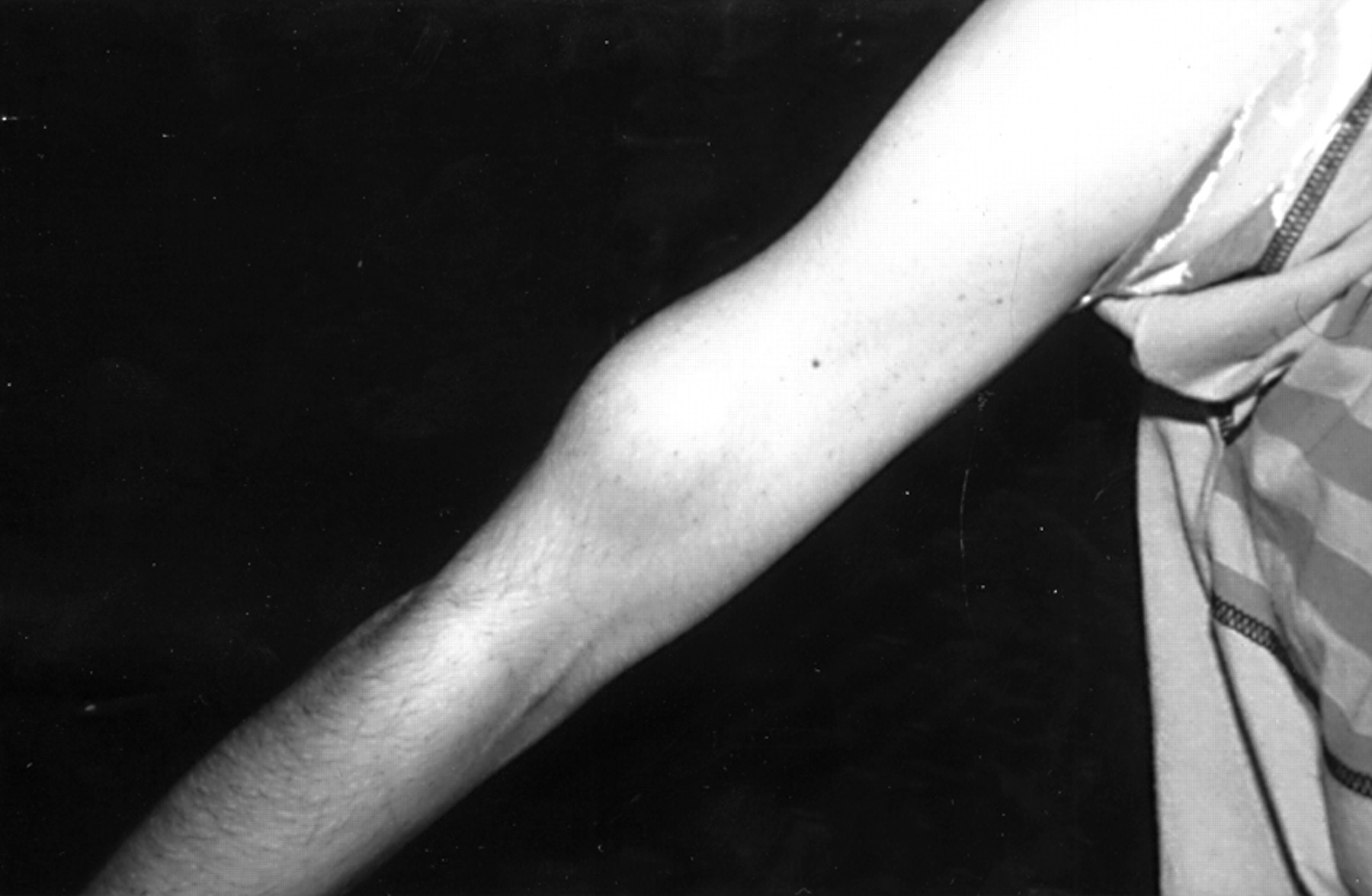
The skin is a useful aid to diagnosis, but cutaneous features in NF2 are much more subtle than in NF1. About 70% of NF2 patients have skin tumours, but only 10% have more than 10 skin tumours.15 25 The tumours appear to be of at least three different types. The most frequent type is a plaque-like lesion, which is intracutaneous, slightly raised, and more pigmented than surrounding skin, often with excess hair (fig 2). More deep seated subcutaneous nodular tumours can often be felt, sometimes on major peripheral nerves. These tumours occur as a fusiform swelling of the nerve with thickened nerve on either side (fig 3). There are also occasional intracutaneous tumours similar to those in NF1. The great majority of these tumours are schwannomas, but occasional definite neurofibromas do occur.
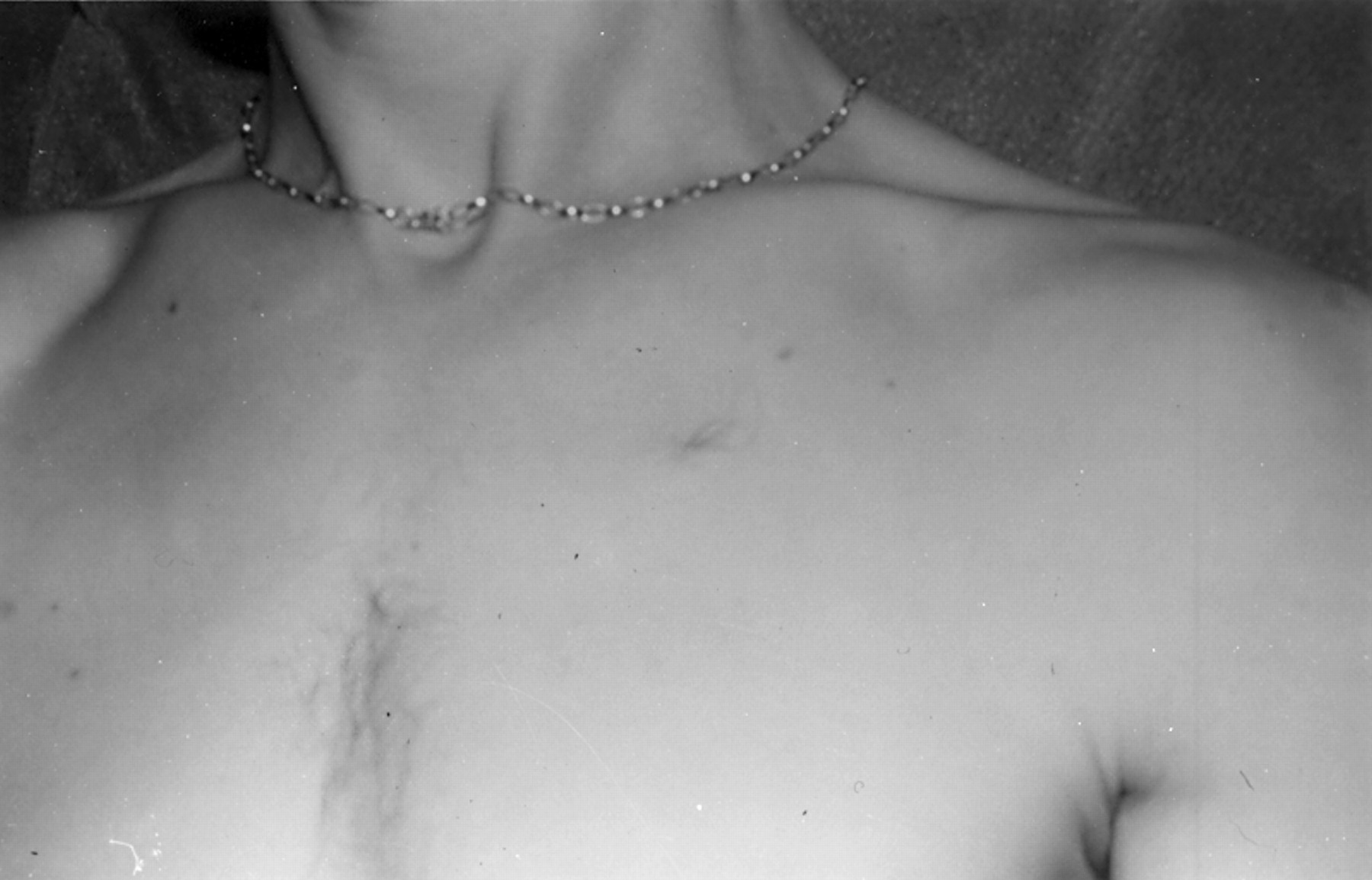
Plaque-like lesions on the chest in a patient with NF2. These are raised, often slightly pigmented lesions that are also frequently hairy.

{kind=link}
{kind=link}
{kind=link}
Subcutaneous schwannoma on a major nerve in the upper arm in a patient with NF2.
OUTCOMES
Even with improvements in microsurgery and with use of radiation therapy, the great majority of subjects with NF2 become completely deaf. The tumours in NF2 are more difficult to treat than those of sporadic unilateral VS, as NF2 VS are often multifocal, appearing “like a bunch of grapes”30 around the vestibular nerve in particular. There is evidence for a histological difference, with NF2 VS being more lobular and less vascular then their sporadic counterparts.31 Patients may also be severely disabled by a combination of poor balance, visual problems, and weakness owing to spinal tumours. Indeed, many NF2 patients become wheelchair bound in early adulthood. Loss of facial nerve function is one of the most feared aspects of the condition for many sufferers, although in good surgical hands this complication is now much less common.32 33 Many patients with multi-tumour disease die in their twenties and thirties.
RADIOGRAPHIC FINDINGS
NF2 can be diagnosed when the criteria in table 2 are fulfilled. The “gold standard” in terms of diagnostic precision is the magnetic resonance imaging (MRI) scan with gadolinium enhancement, which should include a complete spinal scan as well as a cranial scan. MRI will now detect tumours as small as 1-2 mm in diameter on cranial and spinal nerve roots. Many of these small spinal tumours will never lead to symptoms. Full spinal imaging will detect evidence of spinal tumours in 80-90% of patients,34 but older studies that were done before the widespread use of spinal scanning showed that only 25-30% of NF2 patients had symptomatic spinal tumours.15 21 There is also increasing recognition of intramedullary tumours, often associated with a syrinx, which predominate in the upper cervical spine and brainstem. On biopsy these tumours are usually low grade ependymomas. Although these can initially be very worrying for the radiologist or treating clinician, the great majority of these tumours do not progress. Another common finding on MRI is schwannomas on other cranial nerves. These occur most commonly on the 5th nerve,20 but every cranial nerve can be affected in NF2. Nonetheless, it is rare for cranial nerve schwannomas other than VS to grow to a size where removal is necessitated. Meningiomas can easily be detected on MRI as enhanced areas on the meninges around the spinal cord, brain, or optic nerves. These can form confluent areas on scan or “meningioma en plaque”. VS growth rates are extremely variable but average 2 mm per year,35although growth rates are higher in younger patients.36 In contrast, meningiomas characteristically have more rapid growth.
There are several groups of subjects who should be considered at risk and investigated further. These groups include those with a family history of NF2, patients under 30 years presenting with a unilateral VS or meningioma, patients with multiple spinal tumours (schwannomas or meningiomas), and patients with cutaneous schwannomas.21 22 37 MRI scanning is vital in their further assessment.
Although CT scans have been largely replaced owing mainly to their poor sensitivity at detecting small VS,15 a further feature of NF2 can be identified. A proportion of NF2 patients have intracranial calcification from a young age.15 However, this sign is not useful enough to supplement MRI with CT. Without firm clinical justification, repeated CT scans should be avoided in NF2 owing to the tumour prone nature of the disease.38
Pathology
The main tumour sites, their frequencies, and pathologies are presented in table 1. As previously noted, schwannomas can occur at all locations around the body where there are nerves with Schwann cells. The predilection for the superior vestibular branch of the 8th cranial nerve remains unexplained. Schwannomas are encapsulated tumours of pure Schwann cells that grow around the nerve. They may contain blood vessels and have areas of sheets in intertwining fascicles (Antoni A) and looser arrangements (Antoni B).31 The tumours also stain for S-100 protein and vimentin. In NF2, these tumours can be multifocal and have a more lobular architecture than sporadic tumours.31 Spontaneous malignant transformation of these tumours to malignant peripheral nerve sheath tumours (MPNST) does occur, but is more than 10 times as likely to happen after radiation treatment.38 The background rate of 0.5% for CNS malignancy in NF2 is also very much less than for NF1.38 39 A small proportion of nerve related tumours in NF2 are pathologically delineated as neurofibroma. In these tumours there is an admixture of cell types (Schwann cells, fibroblasts, and mast cells) and the tumour usually has identifiable axons within it. Neurofibromas mainly occur in the skin25 (where they are still outnumbered by schwannomas by a factor of 5-10), but also on the spinal nerve roots.40 Halliday et al 40 showed that, in a series of spinal schwannomas and neurofibromas, all spinal tumours in patients with NF1 were neurofibromas while, with one exception, all spinal tumours in patients with NF2 were schwannomas (one patient had a mixed tumour). Neurofibromas are not a feature in the cranium. There is currently no evidence of histological or molecular differences between neurofibromas in NF1 and NF2. Occasionally tumours are found which show features of both schwannoma and neurofibroma, particularly on spinal nerve roots. In contradistinction to NF2, schwannomas and meningiomas do not occur in excess in NF1.39 41
The second most characteristic tumour of NF2 is meningiomas, which usually occur supratentorially in the falx and around the frontal, temporal, and parietal regions. Meningiomas also occur around the spinal cord, and these can be difficult to remove surgically. Although there are different histological types of meningioma (meningothelial, fibroblastic, and transitional), there is no evidence for a clinical subdivision into NF2 related and non-NF2 related meningiomas.42 Collision tumours consisting of a schwannoma and meningioma can occur, particularly in the cerebellopontine angle. Antinheimo et al,13 in a study of all meningiomas and schwannomas in an 11 year period in the Helsinki area, found that 3% of schwannoma patients and 1% of meningioma patients had NF2. A further 2% of schwannoma patients and 4% of meningioma patients had multiple tumours without fulfilling clinical diagnostic criteria for NF2. Nonetheless, the great majority of NF2 patients do not present with an isolated tumour, and there is only a small risk of NF2 after a truly isolated VS (no other features of NF2 on clinical examination or scan).43 However, as many as 10% of those presenting in childhood with an apparently isolated meningioma go on to develop NF2.22
Low grade ependymomas and gliomas occur in NF2 and are now being increasingly recognised. These are very indolent and rarely metastasise around the CNS. The primary location for these tumours is in the cervical spine and brainstem. Malignant progression is sometimes associated with radiotherapy treatment.38
Molecular genetics
Much of the work in isolating the NF2gene involved studies of tumour material. Initially, cytogenetic studies of meningiomas highlighted chromosome 22 as the likely location of the NF2 gene.44 Subsequent cytogenetic studies of schwannomas also confirmed that loss of chromosome 22 or its long arm was by far the most frequent event,45 which was later confirmed by DNA studies.46 Seizinger et al 47 were the first to show loss of constitutional heterozygosity of chromosome 22 DNA markers in tumours from a patient with NF2. Linkage studies then confirmed that all affected members of a large Pennsylvania family4 carried the same copy of chromosome 22.7 The NF2 gene was further localised by the discovery of constitutional deletions in NF2 families, one of which involved the neurofilament heavy chain gene.48 The NF2 gene was then isolated by the simultaneous discovery of constitutional and tumour deletions in a cell membrane related gene termed merlin10or schwannomin.11
Standard mutation techniques, such as single strand conformational polymorphism analysis or denaturing gradient gel electrophoresis, detect between 35% and 66% of causative mutations.49-51Since the majority of these mutations are truncating mutations, leading to a smaller and probably ineffective protein product, more rapid screening techniques such as the protein truncation test can be used. Early studies suggested that missense mutations (which result in a complete protein product) and large deletions (which result in no protein product) both caused predominantly mild phenotypes.48 50 52 Larger studies of detailed genotype/phenotype correlations in multiple families have since been reported.51 53 56 The phenotype is more variable in patients with splice site mutations57; patients with 5′ mutations have more severe disease than patients with 3′ mutations.58 The more severe phenotype in patients with protein truncating mutations may be because of a dominant negative effect, with mutant protein dimerising with the normal product, leaving less wild type protein for tumour suppression.
Some milder cases have mosaic disease,18 59-61 in which only a proportion of cells contain the mutatedNF2 gene. Recent evidence suggests that up to 20% of NF2 cases without a family history of the disease are mosaic, carrying the mutation in too small a proportion of their cells to be detected from a blood sample.18 61 This accounts for the milder disease course in many patients with unfound mutations, and since only a subset of germ cells will carry the mutation, there is less than a 50% risk of transmitting the disease to their offspring.18 However, if an offspring has inherited the mutation, they will be more severely affected than their parent, since the offspring will carry the mutation in all of their cells.60 One of the features that suggested that mosaicism existed in NF2 was that NF2 mutations were harder to find in blood in sporadic cases than in patients who had inherited the disease from an affected parent.18 43 61Mosaicism may be particularly likely in NF2 if the tumours are predominantly on one side.18 The mosaic mutation can be detected by analysing tumour material from an affected subject. If an identical mutation is found in two tumours from that subject, their offspring can be tested for the presence of the mutation.
Further causes (apart from mosaicism) of the low detection rate for mutations in blood using standard techniques are large deletions and rearrangements at the NF2locus.62 However, C>T transitions causing nonsense mutations are the most common mutations in theNF2 gene.50 51 53-56 63
Diagnostic criteria
Constitutional mutations in the NF2gene are found just as frequently in patients who fulfil modified NF2 diagnostic criteria as in sporadic cases with bilateral VS.18 43 This justifies future inclusion of these subjects as having “definite” NF2, rather than having only one criterion (bilateral VS) for “definite” NF2.8 37However, the inclusion of criteria for “probable” NF2 (unilateral VS <30 years plus other NF2 criteria, or two or more meningiomas plus other NF2 criteria37) is a helpful addition.
Management
NF2 presents difficult management issues and patients optimally should be managed by a multidisciplinary team consisting of a neurosurgeon, otolaryngologist, audiologist, ophthalmologist, neuroradiologist, and geneticist. Surgical results are certainly far better when managed by an experienced team.32 33 It is important to balance the use of microsurgery and radiation treatment, which can have a role in patients who have particularly aggressive tumours, or who are poor surgical risks, or who refuse surgery.64 Teams experienced in the positioning of brainstem implants can offer partial auditory rehabilitation to those who are deaf, although results are still behind those achievable for cochlear implants. Although the cochlear nerve may be left initially intact after surgery its blood supply may be damaged; nonetheless a few patients can be rehabilitated successfully with a cochlear implant. Because detection of tumours at an early stage is effective in improving the clinical management of NF2, presymptomatic genetic testing is an integral part of the management of NF2 families.
Predictive diagnosis by linkage analysis using intragenic markers or markers flanking the NF2 gene is now possible in the great majority of families with two or more living affected members.9 65 66 Once a mutation has been identified in an affected subject, a 100% specific test is available for that family. However, mutation detection is time consuming and expensive, and may not reveal the causative mutation. In most families with more than one affected subject, linkage analysis will still remain the test of choice since it will give >99% certainty of affected status. By combining this with a cumulative age at onset curve,17 the risk to an unaffected 30 year old with a normal scan and favourable DNA result is infinitesimally small, although 100% confidence can still be attained only with the identification of family specific mutations. Age at onset curves aid genetic counselling; for example, the risk of having inherited NF2 for an asymptomatic at risk subject 25 years of age, before screening, will have dropped to 25%. As more families are followed prospectively, a curve for age at diagnosis with screening will become available. At risk subjects who are shown not to have inherited the mutatedNF2 gene do not need further follow up.
SCREENING PROTOCOL
Children of affected patients should be considered to be at 50% risk of NF2 and screening for NF2 can start at birth.21Cataracts can affect vision in early life and other tumour implications are present in the first 10 years of life.22 23 28Formal screening for VS should start at 10 years, as it is rare for tumours to become symptomatic before that time even in severely affected families. Annual audiological tests including auditory brainstem response are still a useful adjunct to MRI. Surgery is unlikely to be more successful for tumours <6 mm than for tumours sized 6 mm,17 but VS growth is higher in younger patients,36 so for asymptomatic at risk subjects without tumours, MRI screening every two years for those <20 years old and every three years for those aged >20 years should be sufficient. The initial MRI scan could be at around 12 years of age, or earlier in severely affected families. Once tumours are present, MRI screening should probably be annual. Spinal tumours are found very frequently on MRI, as discussed previously. While only 25-30% of patients with spinal tumours require a spinal operation for a symptomatic tumour,15 21 a full annual neurological examination is probably a wise precaution. In most families it is now possible to develop a genetic test so that screening can be targeted to affected subjects only. Uptake of presymptomatic genetic testing in childhood and adult life is high.67
Differential diagnosis
The main possible diagnostic dilemma with NF2 occurs in isolated patients with multiple non-cranial schwannomas. Some of these patients may well go on to develop NF268 and all require a cranial MRI scan. Some patients can be proven to be mosaic for anNF2 mutation.18 69 There are nonetheless a small group of patients with tumours largely confined to the skin and spine, with sparing of the 8th nerve, who can be proven not to have an underlying NF2mutation.69 These subjects may pass the condition on to their children, and in families where this occurs there is still tight linkage to the NF2 locus,68 69although NF2 mutations are found in only a minority of patients with this variant form of the disease.70 Confusion with NF1 is unlikely since only 1-2% of NF2 patients have six or more café au lait patches15 25 and Lisch nodules are rare in NF2,21 but review of tumour histology is a wise precaution in equivocal cases. The presence of a schwannoma in a patient who does not fulfil NIH criteria for NF18 makes NF1 extremely unlikely, while the presence of multiple neurofibromas makes NF2 very unlikely.
MODIFYING FACTORS
Early reports suggested that NF2 was worse if the disease was inherited from the mother12 14 but, as previously noted, this may be because of decreased genetic fitness in males. In addition, there is no evidence for genomic imprinting on chromosome 22q. It has also been suggested that the disease course is worse in affected females.19 71 However, more recent data indicate that the effect is mainly on meningiomas rather than on schwannoma growth and development.72
On the whole, NF2 disease course does breed true in families.19 54 73 Some families have a mild disease course, with late onset and few if any CNS tumours other than VS. Other families have a more virulent course, with early onset and death owing to multi-tumour disease. The evidence to date of genotype/phenotype correlations is encouraging and may well lend an insight into the disease process. Nonetheless, much will depend on stochastic events, such as loss of the second NF2 allele, since the disease course is not identical even in monozygotic twins.74
NF2 protein
The NF2 protein, termed merlin or schwannomin by the two groups that identified it independently, is a cell cytoskeleton associating protein of 595 amino acids coded by the 17 exons of theNF2 gene. The name merlin derived from the sequence homology and shared overall domain structure with ERM (moesin, ezrin, and radixin) superfamily of membrane cytoskeleton linker molecules.10 Alternative splicing of exon 16 gives rise to two isoforms, which differ by the last C-terminal 11 amino acids. The NF2 protein is expressed in many tissues including neurones, Schwann cells, and meningeal cells. Normally, it exhibits a linker function by binding transmembrane adhesion molecules such as CD44 (hyaluronate receptor) and cytoskeleton components (F-actin, microtubules, β-spectrin).75-77 Accordingly, NF2 protein impairs cell adhesion, motility, and spreading properties, which are known to be essential for tumour formation.79 The NF2 protein binds to ezrin and thus probably associates also functionally with ERM proteins, which have the ability to oligomerise and bind to each other.80 Thus, the NF2 protein is a unique type of tumour suppressor protein with an as yet undefined mechanism of action, which may extend to coordination of adhesion and cell signalling pathways, functions recently shown for ERM proteins.81 ERM proteins associate with Rho and the cAMP dependent protein kinase A (PKA) intracellular signalling pathways and the E-cadherin/β-catenin complex,82 which have a role in control of cell proliferation. Interestingly, also, the NF2 protein binds to Rho-GDP dissociation inhibitor, a component of the Rho signalling cascade.83 Another mechanism may be that the NF2 protein and ERM proteins via their interaction with PDZ domain containing molecules (Na+-H+ exchanger regulatory cofactor, hNE-RF or EBP50) regulate the outside-in signalling events by clustering transmembrane proteins and key components of downstream transduction pathways.84 The genotype/phenotype correlation at the protein level arises from the evidence that nonsense mutations result in unstable protein product, while missense mutations that change only one amino acid may give rise to a protein product with defective negative growth regulation.78 On the other hand, in experimental models, the potential to inhibit cell growth is associated only with isoform I, but not with isoform II nor C-terminal deletion constructs, both unable to form interdomain head to tail interactions, suggesting that the protein conformation may be relevant for the tumour suppressor function.85 However, as yet too little is known about the cellular events that account for theNF2 gene's ability to suppress growth in Schwann cells nor in other cells. Nonetheless, it is now clear that loss of NF2 protein is the main and possibly only rate limiting step in the formation of all schwannomas and most meningiomas.86 87
The future
The much awaited step in NF2 was the localisation and cloning of the gene. This achieved, precise diagnosis is possible if a specific mutation can be found, but this may also allow prediction of the disease course by genotype/phenotype studies. Clinical factors that are closely related to mutation type, such as age at onset or age at diagnosis, may also predict the course of the disease.36 88 This may be particularly useful in patients with new mutations, in whom insight into the likely speed of tumour progression and risk of other tumours would be very helpful. Although DNA predictive testing for 70% of families is available with flanking markers or mutation testing,17 there is currently little demand for prenatal NF2 diagnosis in the UK (three tests in the last six years). A less controversial option would be preimplantation diagnosis, which is being evaluated in a limited sense for other diseases, such as familial adenomatous polyposis and cystic fibrosis.
The real hope is that the discovery of the gene and its transcriptional protein product, merlin/schwannomin, will lead to the development of somatic gene therapy. Intuitively, the prospects for NF2 appear promising owing to the relative lack of phenotypic variation in subjects with the same mutation and the paucity of involvement of other genes in the tumours themselves. Replacement of the tumour suppressor product in the tumours through viral vectors or direct recombination of the NF2 gene, although requiring great advances in our knowledge, could be very rewarding. Use of newer drugs, particularly anti-angiogenesis agents, may also offer promise.
References
Linked Articles
- Correction