Article Text

Abstract
The Y chromosome provides a unique opportunity to study mutational processes within the human genome, decoupled from the confounding effects of interchromosomal recombination. It has been suggested that the increased density of certain dispersed repeats on the Y could account for the high frequency of causative microdeletions relative to single nucleotide mutations in infertile males. Previously we localised breakpoints of an AZFamicrodeletion close to two highly homologous complete human endogenous retroviral sequences (HERV), separated by 700 kb. Here we show, by sequencing across the breakpoint, that the microdeletion occurs in register within a highly homologous segment between the HERVs. Furthermore, we show that recurrent double crossovers have occurred between the HERVs, resulting in the loss of a 1.5 kb insertion from one HERV, an event underlying the first ever Y chromosomal polymorphism described, the 12f2 deletion. This event produces a substantially longer segment of absolute homology and as such may result in increased predisposition to further intrachromosomal recombination. Intrachromosomal crosstalk between these two HERV sequences can thus result in either homogenising sequence conversion or a microdeletion causing male infertility. This represents a major subclass ofAZFa deletions.
- HERV
- AZFa
- infertility
- Y chromosome
Statistics from Altmetric.com
Increasing numbers of genetic disorders are being shown to result from the recombinogenic effect of flanking repeated sequences. Microdeletions, and more recently their reciprocal recombination product, duplications, have been reported to cause neurofibromatosis type 1,1 Smith-Magenis syndrome,2 and Charcot-Marie-Tooth disease, among other diseases.3Unequal exchange between repeats on homologous chromosomes (UCE) or sister chromatids (USCE) are the proposed mechanisms by which these rearrangements may occur. The pathogenic role of dispersed repeats in sponsoring illegitimate recombination, though documented, has been overshadowed by that of longer region specific paralogous duplications.3 Investigations into the length of sequence identity required to initiate homologous recombination in meiosis has led to the concept of a minimum efficient processing segment (MEPS). In mammalian cells, the MEPS is thought to be at least 200 bp in length.4
Human endogenous retroviral sequences (HERV) are a major subclass of dispersed repeats, accounting for about 1% of the human genome.5 The remnants of ancient germ cell retroviral infections that have been multiply transposed after their original integration, partial and complete HERV proviral sequences are widely distributed throughout the genome.6 The degree of sequence divergence between superfamilies of HERV varies markedly and is thought to correlate with the time since major waves of amplification.7 Until now HERV sequences have not been associated with pathogenic illegitimate recombination.
The human Y chromosome has been recently shown to contain certain dispersed repeats, including Alu and HERV sequences, at a significantly higher frequency than the autosomes.8 It has been suggested that deletions mediated by this higher frequency of dispersed repeats may be the major mode of pathogenic mutation on the Y chromosome.9 It has also been suggested that the history of inversions on the Y chromosome that characterised its divergence from the X also results from this higher density of repeats.10 11
The AZFa, AZFb, and AZFc (azoospermic factor) regions on the long arm of the human Y chromosome have been defined by deletion intervals determined by screening azoospermic and oligozoospermic patients with a variety of probes and STSs from the Y chromosome12 (fig 1). These three loci are involved in infertility phenotypes ranging from oligozoospermia to Sertoli cell only syndrome.12 The AZFaregion (primarily associated with Sertoli cell only syndrome) comprises around 1100 kb that have been fully sequenced and contain at least three genes, DFFRY (also known asUSP9Y), DBY, andUTY. 13 UTY has been excluded from involvement in the phenotype, whereas it has been suggested that bothDFFRY and DBY are involved in infertility, with severe impairment of spermatogenesis occurring when the two genes are defective or deleted.13 A de novo point mutation in DFFRY has been described in a single oligozoospermic patient, but microdeletions remain by far the most common pathogenic lesion.14
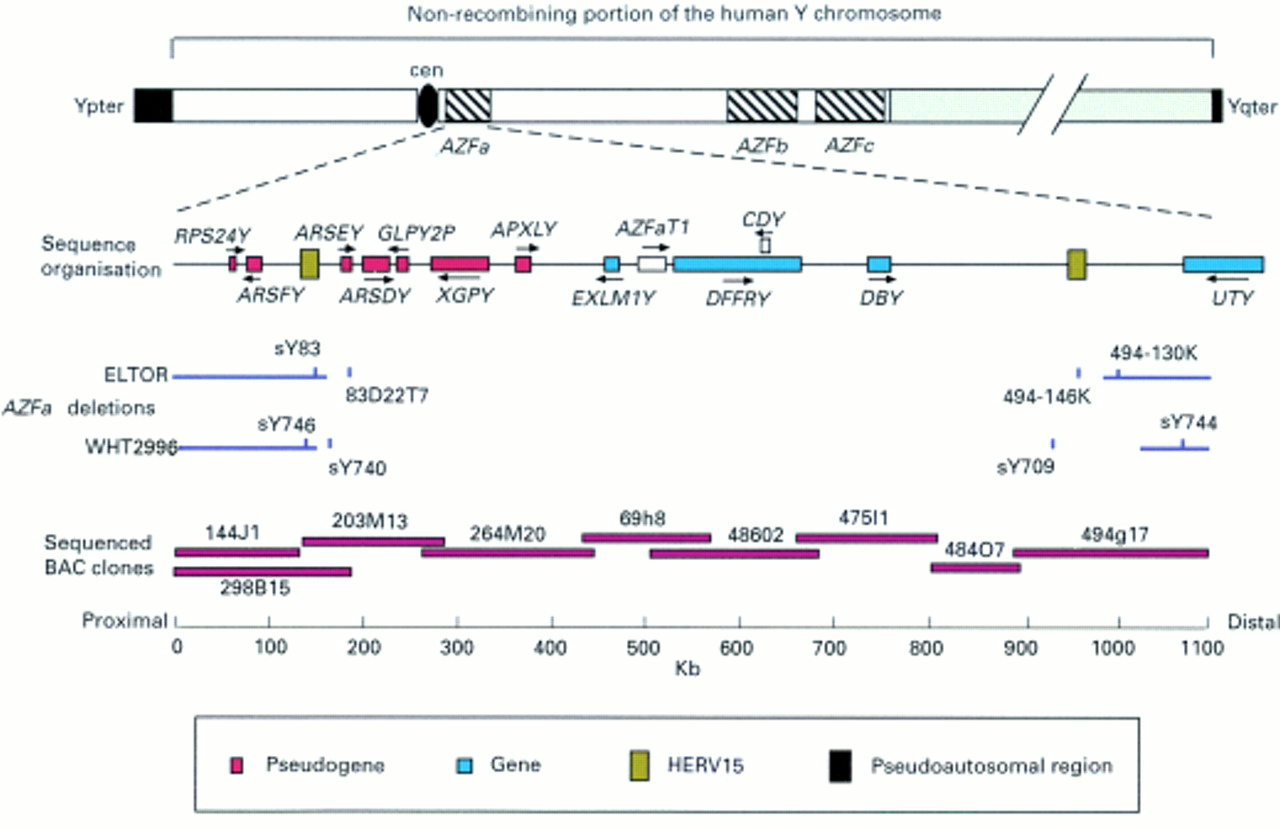
This figure illustrates the AZFa region of the long arm of the Y chromosome. Underneath the labelled ideogram of the human Y chromosome is an expanded view of the sequence organisation of the AZFa region including the genes and pseudogenes mapped previously13 and the HERV15 sequences identified by use of the NIX program (see Materials and methods). Beneath are positioned the previously determined AZFa deletion breakpoints from two patients (ELTOR13 and WHT299614 (see Discussion)) and the STS markers that span the breakpoints. Finally, the organisation of sequenced BAC clones covering 800 kb of the AZFa region14is superimposed on a kilobase scale.
The haploid Y chromosome does not recombine at meiosis over most of its length, and thus it contains within it haplotypes that are changed solely by mutation. Such haplotypes contain a simple record of our evolutionary past and have been exploited to investigate many facets of human population prehistory.15 Non-recombining haplotypes constructed from unique mutations can be used to construct a perfect phylogeny, without reticulation. In the case of the Y chromosome, in contrast to mitochondrial DNA, single nucleotide polymorphisms (SNPs) and certain insertion/deletion polymorphisms (indels) can be considered to be unique on the basis of their producing just such a phylogeny.16 Subsequently, putative recurrences of mutational events can be investigated by mapping them onto this single most parsimonious phylogeny.17 18 In addition, the Y chromosome has proven to be a useful model system for investigating intrachromosomal recombination mechanisms in isolation.19
The first Y linked polymorphism was discovered in 1985, by probing restriction enzyme digests of genomic DNA with a 2.3 kbBglII fragment from the p12f clone.20 From Southern hybridisations ofTaqI and EcoRI digests it was hypothesised that a deletion of roughly 2 kb was the event underlying the polymorphism. The Y chromosomal lineage defined by the 12f2 deletion is of interest for studies of prehistoric migrations and is found at highest frequencies (greater than 25%) in Middle Eastern, southern European, North African, and Ethiopian populations.21
Here we show that a major subclass of AZFamicrodeletions results from a single crossover between two well separated, highly homologous HERV sequences. We also show that a double crossover event between these two HERV copies also underlies the 12f2 deletion, which, contrary to previous thought, can be shown to be recurrent in human evolution. We hypothesise that the sequences generated by these double crossovers may predispose to further single crossovers.
Materials and methods
SEQUENCE ANALYSIS
The BACs containing the deletion breakpoint intervals identified previously in ELTOR (AC002992 and AC005820) were analysed using the NIX programme at the HGMP resource centre (http://www.hgmp.mrc.ac.uk/) and the ENSEMBL database of automatically annotated eukaryotic sequence (http://ensembl.ebi.ac.uk). The consensus sequence of HERV15 was obtained from the latest RepBase update (http://www.girinst.org). Alignments were performed using CLUSTALW (http://www.ebi.ac.uk/clustalw/). Restriction analysis was performed using TACG (http://genzi.virus.kyoto-u.ac.jp/tacg/tacg.form.html) or Webcutter (http://www.ccsi.com/firstmarket/cutter/cut2.html). Nucleotide sequence databases were searched with BLAST 2.0 (http://www.ncbi.nlm.nih.gov/BLAST). Dot plots were produced by the program Dotter at the HGMP resource centre.
PCR
Long PCR was performed on 50 ng of genomic DNA either using the Dynazyme EXT kit from Flowgen or the Extensor long PCR system kit from Advanced Biotechnologies using conditions stipulated by the manufacturer. The primer pairs (MWG-Biotech) used are listed in table1. Cycling conditions used with the Dynazyme EXT kit were 94°C for two minutes followed by 10 cycles of 94°C for 10 seconds, 58°C for 30 seconds, 68°C for 1-1.5 minutes per kb, followed by 24 cycles of 94°C for 10 seconds, 58°C for 30 seconds, 68°C for 1-1.5 minutes per kb plus 20 seconds per cycle, and a final extension step of 68°C for seven minutes. Cycling conditions for the breakpoint PCR with ELTOR DNA using the Advanced Biotechnologies kit were 94°C for two minutes followed by 10 cycles of 94°C for 10 seconds, 58°C for 30 seconds, 60°C for eight minutes, followed by 20 cycles of 94°C for 10 seconds, 58°C for 30 seconds, 68°C for eight minutes plus 20 seconds per cycle, and a final extension step of 68°C for seven minutes.
PCR primers used in this study
Nested PCR to generate sequencing template was performed using the primers in table 1 and 1 U of Taq polymerase (Geneo BioProducts). PCR was carried out in a volume of 20 μl with 1 pmol/μl of each primer, 50 mmol/l Tris-HCl (pH 9.0), 15 mmol.l (NH4)2SO4, 0.1% (v/v) Triton X-100, 2 mmol/l of MgCl2, and 360 μmol/l of each dNTP. The cycling conditions were 94°C for two minutes followed by 14-24 cycles of 94°C for 10 seconds, 58-65°C for 30 seconds, 68°C for 1-1.5 minutes per kb, followed by a final extension step of 68°C for seven minutes. Template for nested PCR was generated from long PCR products by running them on a 0.9% agarose (Geneo BioProducts) gel, cutting out the relevant band and incubating the band in 400 μl of water overnight at 37°C; 2 μl of the eluate were then used in each PCR.
12F2 ASSAY
The 12f2 deletion polymorphism was typed using a newly developed assay. The primers are listed in table 1, and were designed from partial sequence data of the cosmid M13A12, isolated using 12f2 as a probe, from the Y chromosome specific library LL0YNC03. Primers 12f2D and 12f2F generate a specific 500 bp amplicon and primers 3′Sry15 and 3′Sry16 a 820 bp control amplicon which is present in all Y chromosomes. PCR conditions were 33-35 cycles of 94°C for 30 seconds, 59°C for 30 seconds, and 72°C for 45 seconds. The concordance of the PCR assay with the original hybridisation assay was indicated by showing that 23 chromosomes known from previous analysis to carryTaqI/8 kb alleles lacked the 12f2 test amplicon in this assay, whereas 23 subjects known to carryTaqI/10 kb alleles did not. The previously unknown recurrence of the deletion polymorphism was discovered during the screening of a large panel of Y chromosomes which had previously been typed for both the YAP and SRY-1532 polymorphisms (Z H Rosser, M E Hurles, M A Jobling, unpublished observations).
CLONING
Restriction digests were performed withEcoRI from Roche according to the manufacturer's protocol. The ∼9 kb long range PCR fragment spanning the ELTOR deletion breakpoint was gel purified and cloned into pGEM T-EASY (Promega) before sequencing.
SEQUENCING
Sequencing template was generated from nested PCR products using spin columns from Qiagen. All sequencing reactions were carried out using the BigDye terminator cycle sequencing kit from Applied Biosystems according to the manufacturer's protocol. Sequencing products were run on an ABI377 sequencer (PE Biosystems).
Results
We previously localised the breakpoints of theAZFa microdeletion in the patient ELTOR to two tightly defined STS intervals within Yq, on the proximal side between sY83 and 83D22T7 and on the distal side between 494-130k and 494-146k (fig 1). Sequence analysis of these two intervals shows the presence of homologous copies of a human endogenous retroviral sequence (HERV15). The STS sY83 lies towards the end of the proximal copy. The average degree of homology is 94%, but varies markedly along an alignment of the two HERVs, as shown by the dot plots in fig 2. The distal HERV is interrupted by an insertion of L1 material consisting of two smaller L1 fragments lying back to back (fig 3). The presence of 14 bp direct repeats flanking the entire insertion suggests that these fragments have integrated as a unit. The region of the two HERV copies distal to sY83 is particularly highly homologous and we hypothesised that the breakpoint may lie within this region. To test this hypothesis, long range PCR was used to span the breakpoint using primers from the retained flanking STS on either side of the deletion (sY83 and 494-130k). A 9 kb amplicon was obtained. This amplicon was digested with EcoRI to localise the breakpoint further. Five of the six fragments were of the size expected from an in silico digest of the region 3′ to the distal HERV. The remaining 3.1 kb fragment represents the proximal end of this 9 kb amplicon and must contain the breakpoint. This fragment was cloned and fully sequenced. As hypothesised, the breakpoint is perfectly in register within an interval of complete identity between the HERVs of 1242 bp. This interval includes the 3′ portion of the HERV and the 5′ portion of the 3′ LTR (fig 3). As well as removing the genesDFFRY and DBY, the resulting ∼700 kb deletion also removes a single HERV15 copy, a composite of the two original HERVs.

Dot plots of the proximal HERV (X axis) against the distal HERV (Y axis). The LTR15 sequences are shown in grey, HERV15 in black, and the L1 insertion in white. (A) Low stringency dot plot, (B) high stringency dot plot showing regions of greatest homology.

Schematic alignment of HERV15 sequences. The 18 kb sequence encompassing the proximal HERV15 sequence is shown in red, aligned to the 19 kb sequence encompassing the distal HERV15 sequence in blue. Between them, indicated by a dashed line, is the region of homology upon which intervals of identity greater than 100 bp are shown in dark blue, and intervals of identity greater than 200 bp in purple. Positions of the displayed sequences within their BACs are shown at the ends. The STS discussed in the text are shown as vertical green bars. Digestion sites for three restriction enzymes are shown as short vertical black lines. The position of the 13 bp deletion discussed in the text is also shown. Sequence encompassing this deletion is deposited in GenBank under accession number AJ278655. Diagonal lines between the sequences indicate the positions of crossovers: dashed, 12f2 deletion; solid, AZFa microdeletion. The PCR products and sequences discussed in Results are shown below. Sequence is colour coded for which HERV it is specific to and dashed lines indicate regions of identity where sequence cannot be assigned to either HERV. Sequence encompassing the distal conversion breakpoint is deposited in GenBank under accession number AJ278654 and sequence containing the AZFa deletion breakpoint is under accession number AJ278656. PCR primer sequences are indicated by small arrows and their sequences given in table 1.
The 12f2 polymorphism was typed using a newly developed PCR assay, which was shown to be in complete concordance with previous hybridisation analysis.22 A screen of diverse Y chromosomes for the 12f2 deletion using the assay above showed that the 12f2 amplicon was absent from two sets of Y chromosomes with different haplotype backgrounds and geographical locations (data not shown). One of these haplotype backgrounds is defined primarily by the presence of the YAP insertion23 and the derived form of the SRY-1532 polymorphism (also known as SRY10381 of Whitfieldet al 24), the other defined solely by the latter (fig 4). The YAP insertion has been shown to have occurred once in human evolution23 with the ancestral state being the lack of an insertion. The ancestral state of the 12f2 deletion can be inferred to be the undeleted form from the published unambiguous rooting of the Y chromosomal phylogeny.16 25Therefore, the 12f2 deletion must have occurred at least twice during human evolution. This information is summarised in the phylogeny in fig4. This observation of recurrent deletion suggested that an event more complex than a simple deletion underlies the polymorphism(s).

A rooted Y chromosomal phylogeny indicating the recurrent origins of the 12f2 deletion and the lineage affiliations of the two 12f2 deleted subjects sequenced.
The primers used in the 12f2 assay were used in a BLAST search of Y chromosomal contigs that identified the same distal HERV15 sequence identified above. Restriction analysis (in silico) of this sequence showed that the perfect excision of the L1 material described above could generate the polymorphic banding patterns observed in Southern hybridisations of genomic DNA digested with three different enzymes20 (C Tyler-Smith, personal communication). However, no mechanism has been described by which perfect L1 excision can occur recurrently.
Consequently, two overlapping amplicons covering roughly 9 kb of the distal HERV were generated by long range PCR in two subjects, one from each of the two 12f2 deleted Y chromosomal lineages (YAP+, YCC76; YAP−, OXEN). A primer flanking the L1 insertion was used to generate sequence from the more distal of these nested PCR products in each subject. It was found that the L1 material was precisely excised together with one of the flanking 14 bp direct repeats in both 12f2 deleted lineages. In addition, however, sequence 5′ to the site of excision was converted to be identical to the proximal copy of the HERV. There was no conversion of sequence 3′ to the site of L1 excision, thus defining the distal end of the converted sequence in both lineages. Further sequencing was undertaken to isolate the proximal end of the converted sequence. Alignment of the two HERV sequences showed hundreds of base substitutions and indels that differentiate between the two HERV sequences. A total of 238 of these differentiating sites were assayed by sequencing: 112 were found to be proximal to, four distal to, and 122 within the converted sequence. In addition, 3′ to the distal four unconverted differentiating sites lies 110 bp of sequence flanking the HERV that is specific to the distal BAC. The proximal end of the converted sequence was localised to a 1285 bp interval of identity between the two HERV copies in both of the 12f2 deleted lineages, thus defining a minimum intervening interval of 4.6 kb of converted sequence. There was a single difference between the sequences generated from the two 12f2 deleted lineages, a deletion of 13 bp within the converted region of YCC76 (position 144024-144036 within clone 494g17). This deletion could either have occurred after the original conversion event in the recipient HERV or before the conversion event in the donor HERV.
Discussion
AN IMPORTANT SUBCLASS OFAZFa MICRODELETION
Here we define an AZFa microdeletion breakpoint at the sequence level for the first time and add Y chromosomally based infertility to a growing list of diseases resulting from illegitimate recombination between flanking repeats.3Furthermore we show that interchromosomal recombination need not be invoked to explain these events. We note that two recent studies defining AZFa breakpoints have included samples with remarkably similar breakpoints to those defined here.14 26-28 Most strikingly, the proximal breakpoint of the patient WHT299614 lies between the markers sY746 and sY740 (figs 1 and 3). Thus, we conclude that recombination between these HERV copies represents an important subclass ofAZFa deletions.
DOUBLE CROSSOVERS BETWEEN HERV SEQUENCES CAUSE THE 12F2 DELETION
The region of sequence conversion present in the distal HERV copy from subjects from the 12f2 deleted lineages is far longer than gene conversion events observed in any organism, to the best of the authors' knowledge. Thus, we conclude that this sequence conversion results from double crossover events. This mechanism is supported by the positioning of the conversion endpoints in intervals of identity between the HERV sequences longer than 400 bp. In common with other studies,4 the length of identity rather than degree of homology is the more important determinant of homologous recombination. At first sight, the precise removal of a L1 insertion recurrently during human evolution seems highly unlikely. However, the proposed double crossover provides a plausible mechanism by which this can occur.
A MECHANISM OF INTRACHROMOSOMAL RECOMBINATION WITH DIVERGENT OUTCOMES
Exchange of material between the two copies is non-reciprocal as evidenced by the absence of a size change for the fragment from the proximal HERV in Southern hybridisations of 12f2deleted subjects with the 12f2 probe. Thus, recombination would seem to occur because of misalignment between sister chromatids (USCE) rather than the alignment of repeats in cis. In light of this finding, we propose the mechanistic model for recombination between the two HERV shown in fig 5. Crosstalk between these HERV sequences can result in two outcomes, depending on the number of crossovers. Only a single product of each recombinant event shown in fig 5 is observed here. Duplications are difficult to detect using STS analysis alone, but we know that they exist on Yq.17 As yet, the pathogenic effect of duplicating AZFa genes is unknown, though in the light of recent findings with autosomal syndromes2 worth investigating.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A mechanistic model for the divergent outcomes of single and double crossovers between misaligned sister chromatids (USCE). Proximal HERV is shown in white, the distal HERV in black, the L1 inserted material is shown hatched, and the intervening genes are represented in grey.
Counting the number of bands in high stringency filter hybridisations using the probe 12f2 in both TaqI andEcoRI digests (data not shown) suggests that there are between 10 and 30 copies of HERV15 in the genome. Whether there is crosstalk between other copies of this family of dispersed repeats remains to be investigated.
PREMUTATIONS TO MICRODELETIONS?
We note that the two Y chromosomal lineages defined by the double crossover events reported here contain HERV sequences between which there is a substantially longer length of identity (6 kb), which could act as a better substrate for homologous recombination and as such may predispose to AZFa microdeletions. If true, 12f2 deletions could be considered as premutations, analogous to those in triplet repeat disorders.29 Although further work is required to confirm this hypothesis this raises a number of important considerations.
(1) Given the fact that Y chromosomal lineages exhibit greater geographical differentiation than any other locus,30 rates of AZFa deletion may vary substantially between different ethnic groups. Consequently, populations in which the two 12f2 deleted lineages occur at highest frequency should be investigated. Neither of the two 12f2 deleted lineages is found at reasonable frequencies (less than 5%) within the north western European populations on which infertility research has focused.31
(2) If double recombinants can recurrently homogenise sequence between two ∼10 kb repeats on the Y chromosome, the likelihood is that many of the pathogenic microdeletion causing repeats on the autosomes are also homogenised by double recombinant events. The likelihood of these homogenisations being polymorphic on autosomes is higher than for the Y chromosome given that the time since a most recent common ancestor of an autosome is on average four times that of the Y chromosome.15 Thus, the homogenised repeat premutation (HRP) concept can readily be extended to many autosomal and X chromosomal microdeletions.
(3) If single crossovers occur within regions previously homogenised by undetected double crossovers, all breakpoints will appear to have occurred at one end of the homogenised tract. This would produce a false clustering of deletion breakpoints that may be misinterpreted. We recommend that parental repeats of deleted subjects be sequenced to exclude such possibilities when making assertions on breakpoint clustering. Clearly in the case presented here the breakpoint of theAZFa microdeletion lies outside the converted region, and so this is not an issue.
(4) The assumption that markers such as the 12f2 deletion are neutral is required for their use in reconstructing human prehistory. This assumption may be violated if these markers represent premutations, though only if negative selection outweighs drift.32 33
One of the hallmarks of the Y chromosome is the high frequency of amplified repeat sequences distributed throughout the euchromatic and heterochromatic regions. The findings reported in this paper illustrate the importance of flanking repeat sequences that may be disposed to intrachromosomal recombination in creating deletions or insertions. It may be that the majority of deletions of the Y chromosome can be explained by a similar model but involving different repeat sequences. It would be profitable to examine the location of the common breakpoints in AZFb andAZFc patients to assess the involvement of flanking repeats in these deletions.
Acknowledgments
Sequences discussed in the text have been submitted to GenBank with the accession numbers AJ278654, AJ278655, and AJ278656. The authors would like to thank Chris Tyler-Smith for unpublished information and DNA samples. We are grateful to the Y Chromosome Consortium for the supply of genomic DNA and Zoe Rosser for unpublished data. We thank Pieter de Jong for the cosmid library LL0YNC03, constructed at the HGC, LLNL, Livermore, CA 94550, USA under the auspices of the National Laboratory Gene Library Project sponsored by the United States DOE. We would like to thank Professor Tim Hargreaves and Professor Howard Cooke for genomic DNA from patient ELTOR. Maria Shlumukova was funded by a Nuffield Foundation Undergraduate Research Bursary. MAJ is a Wellcome Trust Senior Fellow in Basic Biomedical Science (grant No 057559). This work was supported by the McDonald Institute for Archaeological Research and grants from the Wellcome Trust and the BBSRC. Patricia Blanco was supported by the British Council.
References
Footnotes
-
↵* Present address: School of Biomedical Sciences, Queens Medical Centre, Nottingham, NG7 2UH, UK.