Article Text

Abstract
The Silver-Russell syndrome (SRS) is characterised by severe intrauterine growth retardation, with a preserved head circumference, leading to a lean body habitus and short stature. Facial dysmorphism and asymmetry are considered typical features of the syndrome, although the range of phenotypic variance is unknown. Fifty seven subjects varying in age from 0.84 to 35.01 years, in whom the diagnosis of SRS had been considered definite or likely, were re-evaluated in a combined clinical and molecular study by a single observer (SMP).
In 50 patients the clinical findings complied with a very broad definition of SRS. Notable additional findings included generalised camptodactyly seen in 11 (22%), many with distal arthrogryposis. Thirteen of the 25 males required genital surgery for conditions including hypospadias and inguinal hernia.
Fourteen (36.8%) subjects above school age have received a statement of special educational needs.
Molecular genetic analysis was performed in 42 subjects and has identified maternal uniparental disomy of chromosome 7 in four. The phenotype was generally milder with birth weights for one patient above and three below −2 SD from the mean. Two children had classical facial dysmorphic features, and two had a milder facial phenotype. Of relevance to the possible molecular mechanism underlying this condition, none of the four disomic patients had significant asymmetry.
- Silver-Russell syndrome
- uniparental disomy of chromosome 7
Statistics from Altmetric.com
The original report of Silver et al 1 in 1953 of two low birth weight children with asymmetry and growth retardation was promptly followed by the description by Russell2 of five children with similar features, two with asymmetry. In 1964, Silver3 reported 16 further cases and reviewed the findings of six other published cases. Silver discussed the wide variation in expression of this emerging syndrome and emphasised the distinction of SRS from other causes of poor pre- and postnatal growth. However, neither he nor other authors were able to offer diagnostic criteria for SRS. Of the total of 29 cases, eight were reported to be significantly and one mildly delayed, with no comment on developmental progress made for 10 cases.
Since 1970, the combined term Silver-Russell syndrome (SRS) has been used to describe subjects of low birth weight and reduced postnatal growth, often in combination with asymmetry, café au lait patches, and fifth finger clinodactyly. The classical facial features, as described by Russell, comprise a high forehead tapering to a small jaw. The nasal bridge is prominent, the philtrum well demarcated, and the corners of the mouth downturned (fig 1). Tanner and Ham4reported the use of growth hormone in this condition. The potential for increasing eventual height using early growth hormone treatment is still being assessed.5-7 Postnatal growth in untreated subjects is well characterised.8 9 Bone age is often delayed in early childhood but catches up by puberty, and growth studies in 386 cases found a mean adult height of 151.2 cm (SD 7.8) in males and 139.9 cm (SD 9.0) in females.10

Typical features of a child with the SRS phenotype.
SRS is a well recognised syndrome,11 but diagnostic criteria have remained inconsistent and the range of phenotypic variance unclear. This, at least in part, is likely to reflect the heterogeneous aetiology of the syndrome. A more severe end of the spectrum was described by Donnai et al 12 in 1989, while many milder patients are likely to remain undiagnosed. Most cases are sporadic,13 although Duncan et al 14 described two families with apparent dominant transmission of an SRS phenotype, including asymmetry, over three generations. Additional instances of autosomal recessive and X linked inheritance have also been proposed. Verification of familial examples of SRS has proven difficult, particularly since the facial features of the syndrome tend to lessen with age. Various chromosomal abnormalities have been reported in patients with features suggestive but not characteristic of SRS and include deletions of distal 15q or ring chromosome 15, 18p−, trisomy 18 mosaicism, and diploid/triploid mosaicism. Two subjects with a translocation involving 17q25 have more consistent features of SRS, and a paternally inherited deletion of the CSH1 gene at 17q22-24 has been noted in one subject.15 Genetic changes recorded in SRS have recently been reviewed by Wakeling et al.16
In 1990, Hall17 discussed the possibility of uniparental disomy in SRS following the observation that uniparental disomy of certain chromosomes caused intrauterine growth retardation in mice.17 Uniparental disomy of chromosome 7 was suggested by three cases with unexpectedly severe short stature in association with diseases at this chromosome location. Two had cystic fibrosis caused by homozygosity of a maternal cystic fibrosis mutation, and a third child was homozygous for a maternalCOL1A2 mutation. In addition, postnatal growth failure with an SRS phenotype, but without IUGR, was described by Eggerding et al 18 in a child with maternal isodisomy for 7q and paternal isodisomy of 7p. Kotzotet al 19 reported uniparental disomy of chromosome 7 in three of 25 cases diagnosed with SRS and a further case described as primordial growth retardation. We have ascertained a cohort of subjects diagnosed with SRS to characterise the condition further and review diagnostic criteria. The molecular basis for the majority of cases in this group of disorders remains unclear.
Methods
Following Research Ethics Committee approval, the Child Growth Foundation (a charitable organisation which provides support for patients and their families with a wide range of growth disorders) forwarded a letter of explanation, regarding the study, to all families where a diagnosis of SRS had been suggested. Further patients were referred from growth clinics in London. A single observer (SMP) met 55 families. Using a structured interview, details of the obstetric history, perinatal events, subsequent medical history, and treatment were recorded. Developmental delay or extra educational provision was noted. Fifty-seven subjects were examined. Height, weight, and head circumference was measured. Limb lengths and circumferences were measured using standard methods.20 Venous blood was obtained for DNA21 and for chromosome analysis. Where possible medical notes were obtained and reviewed.
Results
Of the 57 subjects examined, two were low birth weight sibs, one with fifth finger clinodactyly, but both were growing along normal centiles. Five further subjects were excluded, two with alternative diagnoses (hypomelanosis of Ito, Floating Harbour syndrome), two with dysmorphic features inconsistent with SRS but with no suggested alternative diagnosis, and one child with a strong family history of short stature and no dysmorphism. The remaining 50 patients are described in detail, separated into those with classical facial dysmorphism and those non-dysmorphic with much milder facial features. These groups were further divided into those with birth weights above or below −2 SD from the mean (fig 2). They comprise 25 males and 25 females aged 0.84 to 35.01 years. Eight were over 16 years. The average maternal age and paternal age was 27.6 and 30.4 years respectively at the time of the proband’s birth.

Structure used for the analysis of the study group.
Table 1 shows some of the clinical findings. Measurements are those recorded at the time of the study. The centile for head circumference was above that for height in 35 cases, but there were children without sparing of head growth, most notably case 3. This child had striking SRS facial dysmorphism. There was no history of hypoglycaemia and she had a statement of special educational needs. Fig 3 shows 12 cases from within the “classical” group. The photographs show how development of a rounder face in females and jaw growth in males lessens the dysmorphic features with age. Case 4 in fig 3 is the same patient as case 1 reported by Donnai et al.12 He had a very severe phenotype at birth but is now comparable to other children of a similar age.
Summary of salient clinical features of the cohort

Twelve subjects with SRS from the “classical” group. Photographs 1-12 are cases 12, 1, 7, 5, 17, 20, 16, 9, 10, 28, 31, and 8 respectively from table 1. (All photographs reproduced with permission.)
PREGNANCY, PERINATAL, AND PAST MEDICAL HISTORY
From maternal recollection, intrauterine growth retardation was suspected in 29 cases. In 11 cases this was before 26 weeks. One mother was prescribed antihypertensive treatment throughout. Only eight babies were delivered prematurely and this was an elective decision between 31 and 36 weeks because of concern about growth. The mean SDS birth weight of the whole cohort was −2.94. One child required brief ventilation and 29 were admitted to special care, mostly for tube feeding. Severe feeding difficulties, including little interest in food and a requirement for small frequent feeds with hospital admissions, were experienced by 56% of parents. Sweating and pallor in the early weeks at home was described by 52% of parents, equally spread across all groups. This may have represented hypoglycaemia and was not investigated in most cases.
Importantly, 13 (52%) males (all but one in the low birthweight groups) required genital surgery (eight for undescended testes, four for hernia repair, and one for hypospadias). In one case the genitalia were described as ambiguous. One female patient has a bicornuate uterus. Subsequent health in most cases was good although one adult patient has developed diabetes and was also treated for pericarditis. One child had a cleft palate repair and one a unilateral pyeloplasty. Thirteen (26%) had been referred to ENT surgeons with recurrent ear infections and concern regarding hearing. Overcrowding of the lower teeth was a common problem in all children with micrognathia, leading to multiple extractions. One patient had undergone surgery to extend her lower jaw but several patients had limitation of jaw opening.
Twenty four (48%) of the cohort had received growth hormone at some stage. Fifteen were over 5 years at start of treatment (range 18 months to 14 years). Four children had less than a two year trial. Only one child (case 36) had evidence of impaired growth hormone secretion. For patients over 18 years at assessment, the mean height, weight, and OFC SDSs were −3.25 (SD 0.72), −2.75 (SD 1.65), and −2.00 (SD 1.01) respectively. Two affected females have had a total of five unaffected children.
EDUCATION
Overall 38 patients were of school age. Fourteen (38%) have received a statement of special educational needs and four of these attended special school. Looking at children aged over 5 years, there is no significant difference between the mean OFC SDS for children who do or do not require extra help at school. Ten children have had speech therapy.
CLINICAL FINDINGS
Seventeen patients (34%) were asymmetrical, with a limb length discrepancy greater than 0.5 cm. Limb circumference was also affected in all of these cases. All asymmetrical subjects had the classical facial appearance. The lower limb was involved in all cases. Ten also had upper limb asymmetry, and in these truncal and facial asymmetry was more often noticeable. The maximum leg length difference was 2.5 cm in a child under 5 years and this was the only subject referred for possible orthopaedic correction. In older asymmetrical patients, limb length differences were typically 2 cm and the history was of little or no progression from childhood. All adult patients with asymmetry had a postural scoliosis and complained of recurrent back pain.
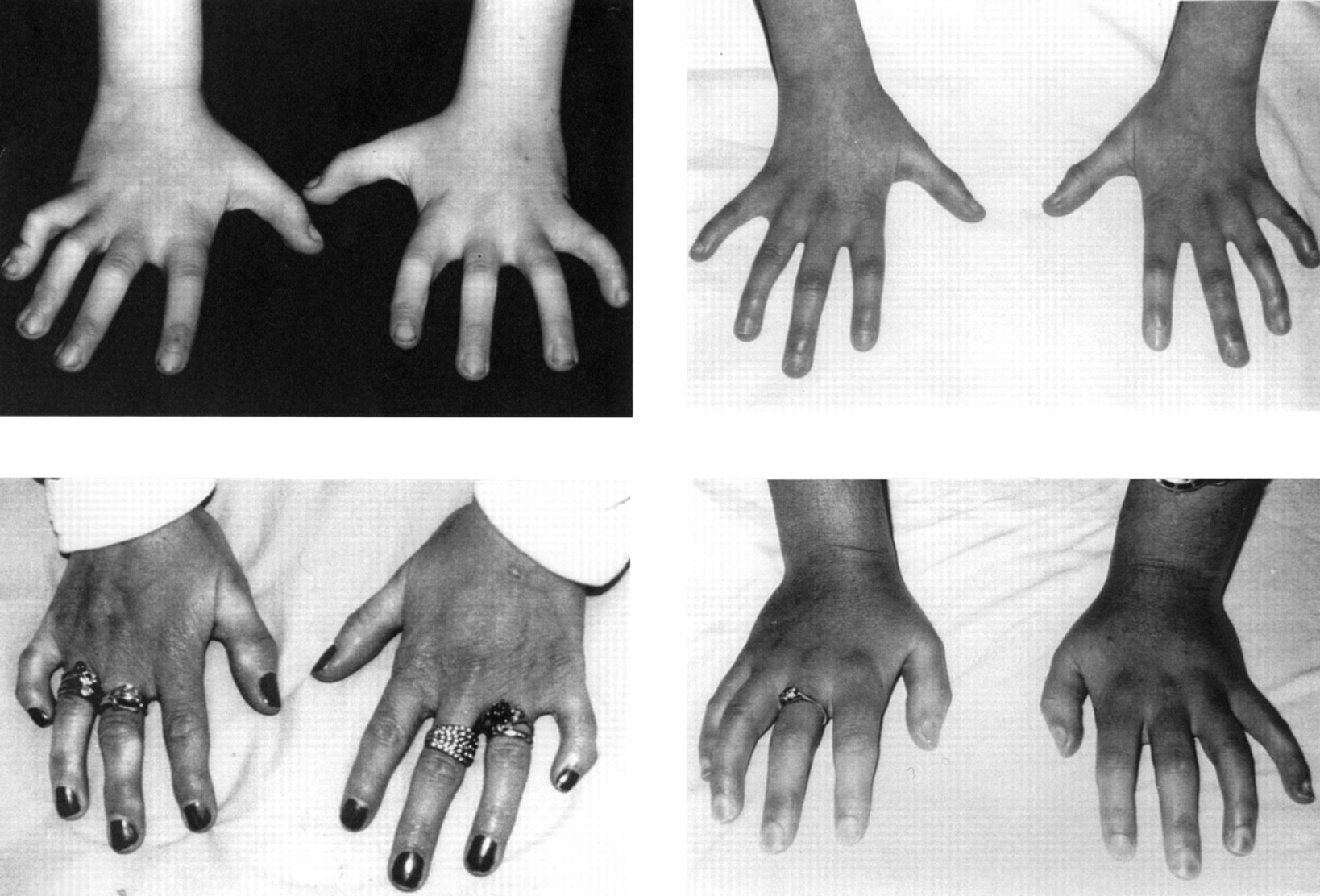
In addition to clinodactyly, or a small fifth finger, 10 of the patients with classical facial features had camptodactyly of all fingers with arthrogryposis of the terminal interphalangeal joints (fig4). This had been noted below 5 years. Nine of these 10 patients were asymmetrical, but the hand features were symmetrical. Three adults describe pain and stiffness in their hands, which appears to be progressive. However, hand radiographs in two adult cases were reported as normal, apart from fifth finger clinodactyly. In addition one patient has limited elbow extension resulting from bilateral dislocation of the radial heads. One patient had a unilateral duplicated thumb.

Camptodactyly and terminal interphalangeal contractures in subjects with SRS.
Two patients had café au lait patches and one vitiligo. Examination was otherwise unremarkable.
FEATURES OF SRS ASSOCIATED WITH UNIPARENTAL DISOMY OF CHROMOSOME 7
As detailed elsewhere by Preece et al,21 42 of these families have been studied for uniparental disomy of chromosome 7. This has been confirmed in four cases (fig 5). Detailed mapping of the disomic regions in these cases and one subsequent case has been described.22 We now report the clinical summaries of the four cases in this cohort (table2) Three had a birth weight below −2 SD and none was asymmetrical. Case 2 in table 2 has a classical facial phenotype and case 1 less marked. However, while cases 3 and 4 have a tall forehead and small jaw they would be considered non-dysmorphic.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Facial features of patients with UPD(7).
Clinical features of UPD(7) cases
CHROMOSOME ANALYSIS
A normal lymphocyte karyotype was obtained in all but one child in the low birth weight/classical/symmetrical group with 47,XXX. There was no evidence of mosaicism.
PARENTAL GROWTH DATA
The mean maternal SDS was −0.41 (SD 0.81) with a mean paternal SDS of −0.34 (SD 1.10) and mid-parental SDS of −0.94 (SD 0.76). This suggests that as a group the mothers were unusually short but not the fathers.
For those subjects who had never received growth hormone, correlating height SDS with parental SDS and birth weight with maternal height showed that the usual relationship between parental and offspring height is lost, and that maternally mediated intrauterine restriction is not part of the problem. This is unlike the pattern in other growth disorders such as Turner syndrome and skeletal dysplasia where parental height has an influence on final height outcome.
Discussion
SRS has a range of phenotype from mild to severe and may well be a heterogeneous group of conditions. Until the paper by Laiet al 23 in 1994 there had been debate regarding the incidence of learning disability in SRS. Silver had suggested that the incidence may be raised but this was not confirmed in subsequent cohorts studied for growth or stressed in later descriptions of the syndrome. For the 25 subjects studied by Laiet al, 23 32% scored within the learning disability range, assessments of special educational needs had been completed on 36%, and IQ score correlated with OFC. The frequency of learning disability in our group is comparable although we cannot find a correlation with OFC, feeding difficulty, or possible hypoglycaemic symptoms. Subjects with learning disability were seen in all phenotypic groups. One reason why the confirmed incidence of learning disability may be under-reported is that many parents state that the performance problems, particularly in reading and mathematics, was not apparent until mid to late childhood. At younger ages it is also likely that observers may have reduced expectations of a child of similar size to a much younger age group. Except in cases with very low birth weight and marked cranial sparing, motor milestones were not delayed and concern regarding development was only raised in four cases at school entry.
Criteria for diagnosis of SRS have not been established. Laiet al 23 diagnosed SRS when three of the following were present: (1) low birth weight (at least 2 SD below the mean adjusted for maternal stature, gestation, ordinal position of the child, and gender), (2) short stature at the time of diagnosis, (3) a characteristic craniofacial appearance as described by Russell, (4) limb, body, or facial asymmetry, and (5) clinodactyly. Within our cohort there was a more homogeneous subgroup defined by classical facial dysmorphic features with a higher frequency of asymmetry and hand anomalies. The pain and stiffness of terminal interphalangeal joints has not been described before, and again in all but one case was with a classical facial phenotype. Maternal UPD(7) was not found in this group. The major features in this subgroup form useful criteria when considering a diagnosis of SRS: birth weight below or equal to −2 SD from the mean; poor postnatal growth, below or equal to −2 SD from the mean at diagnosis; preservation of OFC; classical facial phenotype; and asymmetry.
Our homogeneous group generally had at least four of these criteria. Six cases with classical facial dysmorphic features had birth weights above −2 SD from the mean (including three asymmetrical cases and one child with maternal uniparental disomy). It is difficult to sustain a diagnosis of SRS at higher birth weights but there were typical cases in our cohort, particularly if birth weight was well below that of sibs or when compared to parental size. In 21 of the 31 cases in this group the OFC was above −2 SD from the mean. Subjects with smaller OFCs were in the lowest birth weight group and there was still cranial sparing in most cases. A further subsection had an appropriate growth pattern, a triangular facies, with or without clinodactyly, but including a typical feeding history. It may be more useful to reserve the term SRS for these more stringent groups only.
The low birth weight child who is non-dysmorphic with a prominent forehead and triangular face is more likely to be diagnosed as SRS if they have fifth finger clinodactyly, which in itself is not uncommon. This seems to be the main feature responsible for expanding the phenotype. Case 4 in fig 4 is typical of this group. She did have typical feeding problems with possible hypoglycaemic symptoms; however, UPD case 3 had none of these problems. In these cases it is more important to ask carefully about feeding pattern and sweatiness/irritability relieved by feeding. It may be that UPD(7) will be found more frequently in cases where SRS is considered but may be discounted if stricter criteria are used. The eight cases at the bottom of the table that would be excluded very much reflect the overlap between SRS and IUGR.
We would hope that increased understanding of the molecular mechanisms involved will begin to separate patients within this heterogeneous group.
Acknowledgments
We would like to thank the Child Growth foundation for their support and administrative help, Serono Laboratories who funded travel expenses, and all referring paediatricians and clinical geneticists for their cooperation.