Article Text

Abstract
We report on a boy with a maternal uniparental disomy for chromosome 14 (UPD(14)). At 7 years of age he was referred to us by the paediatrician because of symptoms of Prader-Willi syndrome (PWS). He showed short stature, obesity, mild developmental delay, cryptorchidism, and some mild dysmorphic features. The history further indicated intrauterine growth retardation at the end of the pregnancy. His mother was 44 years of age at the time of his birth. After birth he showed hypotonia with poor sucking, for which gavage feeding was needed. Motor development was delayed. After 1 year he became obese despite a normal appetite. Recurrent middle ear infections, a high pain threshold, and a great skill with jigsaw puzzles were reported. There were no behavioural problems or sleep disturbance. Chromosomal analysis was normal (46,XY). DNA analysis for Prader-Willi syndrome showed no abnormalities. Two years later he was re-examined because we thought his features fitted the PWS-like phenotype associated with maternal UPD(14). At that time precocious puberty was evident. DNA analysis showed maternal heterodisomy for chromosome 14. In all the previously described 11 cases with maternal UPD(14), a Robertsonian translocation involving chromosome 14 was detected cytogenetically before DNA analysis. This is the first report of diagnosis of maternal UPD(14) based on clinical features. This finding underlines the importance of DNA analysis for maternal UPD(14) in patients with a similar PWS-like phenotype even without previous identification of a Robertsonian translocation involving chromosome 14.
- uniparental disomy
- chromosome 14
- genomic imprinting
- Prader-Willi syndrome
Statistics from Altmetric.com
Uniparental disomy (UPD) is the presence, in a diploid subject, of a chromosome pair that derives from only one parent.1 After the first report of a patient with cystic fibrosis and very short stature because of the presence of two identical copies of a maternal chromosome 7 in 1988,2during the last decade 29 of the 47 possible uniparental chromosome pairs (including the XY pair) have been identified.3Clinical effects of uniparental disomy have been reported for several chromosomes.4 ,5 Eleven patients with a maternal UPD for chromosome 14 have been reported so far.6-15 A distinct maternal UPD(14) phenotype is emerging from these descriptions. Intrauterine growth retardation (IUGR), followed by postnatal hypotonia, resulting in feeding problems and motor delay, short stature, obesity, precocious puberty, and mild mental delay are the main features of this condition. Dysmorphic features (mainly of the face and hands) have been described but are not very specific. An overlap with Prader-Willi phenotype was suggested by Berends et al.15 In all patients published so far, DNA analysis for UPD(14) was performed after cytogenetic analysis showed a Robertsonian translocation (13;14, 14;14, 14;21).15 There are two prenatal reports on maternal UPD(14).16 ,17 In both cases, prenatal diagnosis was performed because of maternal age. Cytogenetic analysis showed mosaic trisomy 14 and DNA analysis showed a maternal UPD(14) in the disomic cell line.
We report on a boy with a maternal UPD(14) who has a normal karyotype. DNA analysis for maternal UPD(14) was performed because of his PWS-like phenotype. A review of the clinical features of patients with maternal UPD(14), including our patient, is given in table 1.
Clinical data of patients with maternal UPD(14)
Case report
The proband, a 9 year old boy, is the second child of healthy, unrelated parents. His mother was 44 years of age at the time of his birth. At the end of the pregnancy intrauterine growth retardation was noted. Delivery was at term by caesarean section because of fetal distress. Birth weight was 2250 g and length was 43 cm (both below the 3rd centile). He needed gavage feeding for three weeks because of poor sucking. He was described as a quiet, passive, and friendly baby who never cried. He was hypotonic and his motor development was delayed. He sat at 12 months. At first length and weight progressed along the 3rd centile but after 1 year his body weight increased rapidly and was soon above the 97th centile. His appetite was normal. Middle ear infections were frequent. His pain threshold was considered to be high. There were no behavioural problems or sleep disturbance. Mental development was slightly delayed. He has great skill with jigsaw puzzles. Between 3 and 5 years of age there were periods with hypoglycaemia and a diagnosis of hyperketotic hypoglycaemia was made. Because of his unexplained obesity and small stature he was examined for endocrinological abnormalities, but none was found.
At 7 years 7 months he was referred to us by the paediatrician on suspicion of having Prader-Willi syndrome. Length was on the 3rd centile and body weight above the 97th centile. Head circumference was on the 50th centile. Some dysmorphic features were noted, including a short nose with anteverted nares, downturned corners of the mouth, prominent central incisors, tapering fingers, clinodactyly of the fourth and fifth fingers, and a simian crease on the left hand. His penis was small but not too short. Testes were palpated high up in a hypoplastic scrotum.
Chromosomal analysis and DNA analysis for Prader-Willi syndrome showed no abnormalities. Additional investigations were performed to exclude fragile X syndrome, hypochondroplasia, Bardet-Biedl syndrome, and Cohen syndrome. DNA analysis for fragile X syndrome was normal. X rays showed a delayed bone age, but no other abnormalities. Ultrasound studies of the kidneys were normal, as were ophthalmological investigations.
Recently we contacted the parents again because we thought his features might fit the “maternal UPD(14) phenotype”. Obesity was still a major problem (fig1). His height had accelerated since the age of 8 years and was now between the 25th and 50th centile. Weight was still well above the 97th centile. Precocious puberty was noted. Secondary sexual characteristics were P3G2 (Tanner stage). The testes were in the scrotum now and measured 6 to 8 ml. He had hearing problems because of recurrent otitis media. He was attending a special school for children with hearing and learning difficulties. He was very eager to learn, but found arithmetic rather difficult. His IQ was around 90-100.

Proband at 9 years.
LABORATORY INVESTIGATIONS AND RESULTS
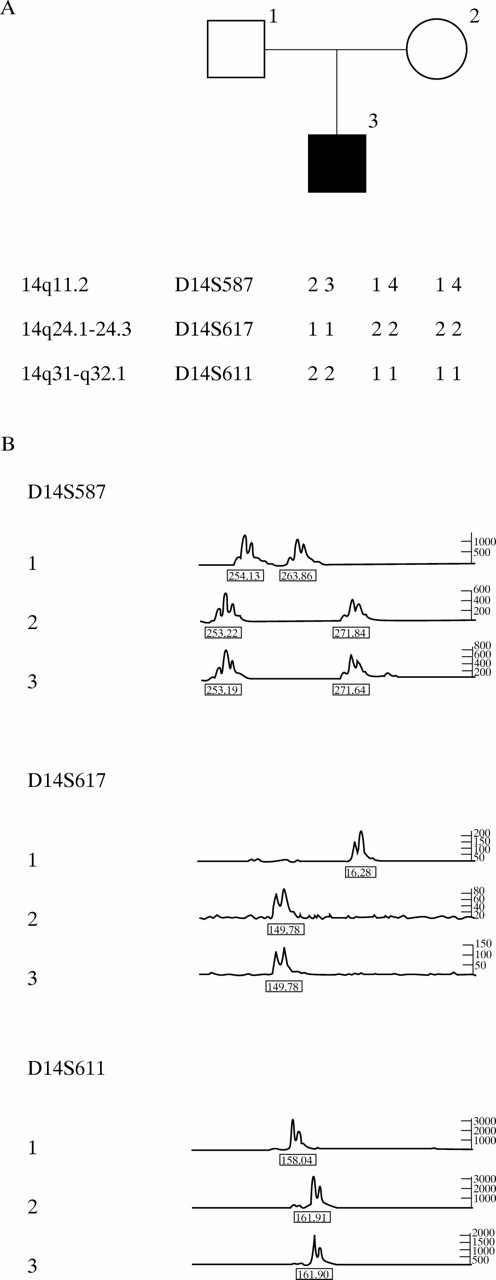
The parental origin of chromosome 14 in the proband was determined by analysing the genotypes of microsatellite markers in the peripheral lymphocyte DNA of the patient and his parents. The informative PCR markers used were D14S587, D14S617, and D14S611. The results indicated that the proband had inherited two alleles from his mother and none from his father. The results of marker D14S587 indicated maternal heterodisomy for chromosome 14 (fig 2). No indication of non-paternity was observed.

{kind=link}
{kind=link}
(A) Chromosome 14 haplotypes in the father, mother, and proband. The relative location of markers at chromosome 14 is indicated. (B) Genotypes of chromosome 14 specific markers (D14S587, D14S617, and D14S611), showing absence of a paternally derived allele (1=father, 2=mother, 3=proband).
Discussion
The maternal heterodisomy for chromosome 14 in our patient is most probably the result of a non-disjunction error during maternal meiosis leading to a trisomic zygote, followed by loss of the paternal chromosome 14 (trisomy rescue). His mother was 44 years of age at the time of his birth. Maternal age is a well known risk factor for trisomy formation.
Clinical effects of UPD can be caused by (1) chromosomal trisomy of the placenta or fetus or both, (2) underlying autosomal recessive disease owing to a reduction to homozygosity in cases with isodisomy, and (3) biallelic expression (or absence of expression) of imprinted genes on that chromosome. When a clinical feature is consistent in many patients with UPD for that particular chromosome, imprinting effects are more likely to be the cause.
The clinical features of all patients with a maternal UPD(14) are summarised in table 1. Intrauterine growth retardation was reported in nine patients. An appropriate weight for gestational age was reported by Temple et al 6 and Antonarakis et al.8 Both children were born at 32 weeks of gestation. Birth length and weight of the girl reported by Coviello et al 12 were not reported. Robinsonet al 9 reported a boy with IUGR who was born at 36 weeks of gestation. These findings suggest that IUGR in maternal UPD(14) is related to the last months of pregnancy. Miyoshi et al 14reported a boy with IUGR born at 29 weeks of gestation, but he was one of triplets.
Premature birth was reported four times, but twice a child was born by caesarian section6 ,14 and in one case8delivery was induced because of rhesus incompatibility. Thus, spontaneous premature birth does not seem to be associated with maternal UPD(14).
Hypotonia was reported in nine patients, often followed by feeding problems (6) and motor delay (9).
Short stature was mentioned in nine cases. Some reports do not contain detailed information on growth parameters, but the available data suggest that short stature can be seen in young children, although it is more obvious in adolescents and adults, probably mainly owing to precocious puberty or scoliosis or both. In seven cases some degree of obesity was mentioned. The age of onset is comparable to that of children with PWS, but the overweight does not seem to be as severe as it can be in children with PWS. Precocious or early puberty seems to be a major feature of this condition. It was reported in all patients of 8 years and older (6). Mental delay, if present (7), was often mild. Dysmorphic features (usually of the face and hands) were noted several times (9), but were not very consistent. Less frequent observations were recurrent otitis media (4), scoliosis (5), and (spontaneously arrested) hydrocephalus (3).
A report of a normal phenotype in a case with maternal UPD(14) can be considered strong evidence against a major imprinting effect. There are two reports in which a normal phenotype associated with maternal UPD(14) was mentioned, but in both cases the evidence is not completely convincing. Morichon-Delvallez et al 17 reported a case where maternal UPD(14) was diagnosed prenatally after finding confined placental mosaicism for trisomy 14. At birth, the clinical examination was normal. Further evaluation, however, is lacking. Papenhausen et al 18 reported on a case of maternal UPD(14) in a woman with a normal phenotype. DNA analysis, however, was not convincing because her father was unavailable for analysis.19 ,20
The maternal UPD(14) phenotype is a recognisable phenotype. Although most cases were assessed after finding a Robertsonian translocation of chromosome 14, our case underlines that maternal UPD(14) can also be suspected solely based on clinical grounds and in the presence of a normal maternal karyotype. Patients with an abnormal phenotype and a Robertsonian translocation for chromosome 14, as well as patients with a Prader-Willi-like phenotype and normal outcome after DNA analysis for PWS, should be re-examined for maternal UPD(14).