Article Text

Abstract
Background Inherited retinal degenerations (IRDs) are a common cause of visual disturbance with a high clinical and genetic heterogeneity. Recent sequencing techniques such as whole exome sequencing (WES) contribute to the discovery of novel genes. The aim of the current study was to use WES data to identify large deletions that include at least one exon in known IRD genes.
Methods Patients diagnosed with IRDs underwent a comprehensive ophthalmic evaluation. WES was performed using the NimbleGen V2 paired-end kit and HiSeq 2000. An analysis of exon coverage data was performed on 60 WES samples. Exonic deletions were verified by ‘PCR walking’ analysis.
Results We analysed data obtained from 60 WES samples of index patients with IRDs. By calculating the average coverage for all exons in the human genome, we were able to identify homozygous and hemizygous deletions of at least one exon in six families (10%), including a single-exon deletion in EYS, deletions of three consecutive exons in MYO7A and NPHP4, deletions of four and eight consecutive exons in RPGR and a multigene deletion on the X-chromosome, including CHM. By using PCR-walking analysis, we were able to identify the borders of five of the deletions and to screen our set of patients for these deletions.
Conclusions We performed here a comprehensive analysis of WES data as a tool for identifying large genomic deletions in patients with IRDs. Our analysis indicates that large deletions are relatively frequent (about 10% of our WES cohort) and should be screened when analysing WES data.
- Clinical genetics
- Vision research
- inherited diseases
- retinal degeneration
- whole exome sequencing
Statistics from Altmetric.com
Introduction
Mendelian diseases in humans can be caused by a variety of different mutation types with missense, nonsense, splice-site and small insertions/deletions being the most common ones. It is relatively straightforward to identify such mutations using classical sequencing techniques. Other mutations, and mainly genomic rearrangements or copy number variations (CNVs), require different detection strategies. Amplification of the DNA of an individual who is heterozygous for such a deletion will result in the amplification of the wild-type allele only leading to a mistakenly concluding result. In special cases, dedicated methods have been developed for the identification of such heterozygous deletions, with multiplex ligation-dependent probe amplification and CNV arrays being the most commonly used. It is therefore reasonable to assume that the current estimated fraction of large deletions (about 7.5%—12 565 out of 166 768 mutations, http://www.hgmd.cf.ac.uk/ac/hahaha.php and ref. 1, 2) is an underestimation.
During the last 5 years, whole exome sequencing (WES) has become a powerful and versatile tool for the identification of various disease-causing mutations.3–6 Its efficacy is especially augmented over gene-by-gene Sanger sequencing and the candidate gene approach7 in genetically heterogeneous diseases such as retinitis pigmentosa (RP) and congenital hearing loss. However, WES analysis fails to detect mutations in many families with hereditary diseases and an average detection rate of only 50–60% has been reported in various conditions.8 Moreover, WES analysis is not efficient for the identification of large deletions as a function of its design, mainly since the traditional WES analysis schemes are designed to identify single-nucleotide variations or those involving a few bases only. Large deletions are not detected by this analysis and one needs to manually inspect exon coverage data of a suspected gene aiming to identify such large deletions.
Inherited retinal degenerations (IRDs; OMIM #268000) are a common group of visual disturbance diseases with estimated prevalence of 1:4000.9–11 These diseases are highly heterogeneous (both clinically and genetically) with autosomal-recessive (AR; 50–60%), autosomal-dominant (AD; 30–40%), X-linked (XL; 5–15%) and mitochondrial inheritance patterns.12 Up to date, more than 250 genes were associated with different phenotypes of IRDs (https://sph.uth.tmc.edu/RetNet/home.htm). Clinically, IRDs can affect diverse functions of the visual system: constricted visual field (RP, rod-cone dystrophy), cone-dominated phenotypes (cone dystrophy, cone-rod dystrophy and achromatopsia), decreased visual acuity with central scotoma (Stargardt disease and Best disease) and nyctalopia (congenital stationary nocturnal blindness, fundus albipunctatus and retinitis punctata albescens). This clinical damage diversity is compatible with the different affected cells within the retinal layers. Generally, the visual damage of most IRDs is a combination of the previously mentioned visual disabilities. In about one-third of cases, retinal degeneration appears as part of other systemic damages named syndromic retinal degenerations such as: Usher syndrome (USH; hearing loss with/without vestibular areflexia), Bardet–Biedl syndrome (obesity, kidney insufficiency, mental retardation and polydactyly) and others.
Lately, mutations in genes that are already known to cause IRDs as well as novel IRD-related genes were found using WES and whole genome sequencing.13–17 WES was found to be useful in the identification of the disease-causing mutations in about 50% of the investigated IRD families.18–22
In the current study, we focus on the identification of homozygous and hemizygous large genomic deletions using WES coverage data analysis. We show that WES is an excellent tool for the identification of such deletions in various IRD-causing genes.
Methods
Subjects and clinical evaluation
The tenets of the Declaration of Helsinki were followed and prior to donation of a blood sample, a written informed consent was obtained from all individuals who participated in this study (see online supplementary figure S1), after explanation of the nature and possible consequences of the study. The research was approved by the institutional review board (IRB) at the Hadassah Medical Center. DNA was extracted from the index patients as well as from other affected and unaffected family members using the FlexiGene DNA kit (Qiagen).
Supplemental material
Ocular evaluation included a comprehensive ophthalmologic exam, Goldmann perimetry, electroretinography, colour vision testing using the Ishihara 38-panel and Farnsworth-Munsell D-15 tests, colour and infrared fundus photos, optical coherence tomography and fundus autofluorescence (FAF) imaging performed as previously described.22
Genetic analyses
Homozygosity mapping was performed using whole genome single-nucleotide polymorphism microarrays (Affymetrix 6.0) as reported previously.23 WES analyses were performed on DNA samples of 64 individuals at Otogenetics (Norcross, Georgia, USA) using different approaches and were grouped accordingly: group 1 (G1)—43 samples using Agilent V4 and Illumina HiSeq 2000 with a 30× coverage (hg19), G2—10 samples using Numblegen V2 and PE100 Illumina HiSeq 2000 with a 30× coverage (hg19), G3—4 samples using Agilent Human exome V5 and PE100 HiSeq 2000/2500 with a 50× coverage (hg19), G4—4 samples using Agilent Human exome V5 and PE100 HiSeq 2000/2500 with a 50× coverage (hg38) and G5—3 samples using Agilent Human exome V5 and PE100 HiSeq 2000/2500 with a 100× coverage (hg19). Sequence reads were aligned to the human genome reference sequence and variants were viewed using the DNAnexus software package. WES data were analysed for potential disease-causing mutations as previously reported.16
Exome coverage analysis
We created an exon coverage database using SQLserver 2014 that includes the following columns: individual number, chromosome number, gene name, start and end nucleotide position of each exon and the mean exon coverage of an individual (MECI). The latter was calculated as the mean coverage values for each base (the number of reads covering a specific nucleotide) in a specific exon. MECI values were averaged in each group of patients (G1–G5) to obtain the mean exon coverage per group (MECG). All groups were analysed with the same set of parameters and filters. Exons with a relatively low MECG (less than mean MECG—SD) were excluded from the analysis. As Y-chromosome-specific exons are not present in females and therefore represent a population of exons that can be used to determine the baseline coverage of total exons that are thought to be deleted, we used the mean coverage data of exons located on the Y-chromosome in 15 female samples. To ensure the inclusion of Y-chromosome-specific sequences, we excluded from the analysis genes that are located on X–Y shared regions as well as genes with close paralogues in the human genome. To allow meaningful mathematical values to be obtained, MECI values of ‘0’ were replaced by a value of ‘0.1’ for calculating the MECG/MECI ratio. Aiming to determine a reliable threshold for the MECG/MECI ratio, the ratio of the 89 suspected exons was plotted and divided into two subgroups based on the slope of the regression line: the first set (including over 50% of the exons) included exons with ratios between 3.2 and 9.9 with a slope of 0.09. On the other hand, the second set with a minimal ratio of 16 showed a much more spaced distribution with a slope of 13.96. We used this change in the regression line equation to select a value that will differentiate these two groups and therefore is likely to include as much as possible true candidates for exon deletion.
Primer design
Primers for the determination of deletion borders were designed using the Primer3 software, and Sanger sequencing of PCR products was used to verify the mutation and to screen additional patients and controls. PCR-walking analysis was performed by designing serial PCR primers covering the suspected borders areas, aiming to identify the borders using the minimal number of PCR reactions.
Sequence analysis
Repetitive elements were screened using the RepeatMasker tool at http://www.repeatmasker.org with default parameters. Palindromic sequences of at least six bases were searched using http://www.biophp.org/minitools/find_palindromes/.
Results
As part of our long-term goal to identify the cause of disease in patients with IRDs, we performed WES analysis on 64 samples (60 IRD patients and 4 controls). We have recently reported that WES traditional analysis (searching for single-base substitutions as well as small insertions and deletions) revealed the identification of disease-causing mutations in known IRD genes in about one-half of cases.22 Aiming to improve WES analysis and identify homozygous and hemizygous large deletions in this set of samples, we tabulated and analysed the mean coverage for each exon in the human genome (n=185 636–194 955 exons, depending on the genome reference sequence version). As can be seen in figure 1A (depicting data of patient MOL0028-4 as an example), the MECI is highly variable. The lower tail of the distribution (red box, figure 1A) contains a few hundred exons that are likely to represent mainly exons with poor coverage (eg, non-coding untranslated exons, polymorphic large deletions and potentially a few pathogenic deletions of exon(s)). We subsequently analysed a total of 106 Y-chromosome exons in females and found a mean MECI of 0.323±0.53 (data obtained from 1590 data points ranging from 0 to 3.3). This set of exons represents the mean MECI values of missing exons and therefore can be used to determine the threshold for the identification of exons that are deleted in a homozygous or a hemizygous state. The filtering stages and their effect on the number of suspected deleted exons are detailed in online supplementary table S2. Based on these values, we analysed base coverage data of exons from 226 known IRD-causing genes.22 We excluded from the analysis exons with a relatively low MECG (lower than 9.6 which is based on mean-SD) across all samples. In addition, we filtered-out exons with a MECI value above 3.3 in a specific individual.

Identification of possible homozygous and hemizygous deletions. (A) The MECI (mean exon coverage of an individual) graph of patient MOL0028-4 showing a large variability in exon coverage ranging from 0 to 200 (a few exons that had extremely high MECI signals and are irrelevant for this analysis were excluded from the graph). The red box highlights exons with a very low coverage ranging from 0 to 3.3 reads per base and therefore might represent exons that are deleted. (B) A schematic representation of the suspected deleted exons in all studied whole exome sequencing (WES) samples. A plot of the ratio between mean exon coverage per group (MECG) and MECI in exons that passed the filtering steps as potentially deleted in inherited retinal degeneration (IRD)-causing genes. Exons are shown for patients in whom no other IRD mutations were detected. Exons exceeding the threshold value of a ratio of 10 are marked in red. The corresponding genes are highlighted above the bar plot. Chromosomes in which such potential exons were identified are marked below the bars. (C) A plot of the ratio between MECG and MECI of exons located on chromosome X (84–96 Mbp) in family MOL0102. Red bars represent exons of the corresponding gene (depicted below the bar plot).
The analysis of all studied WES samples revealed a total of 89 exons that passed the above-mentioned filtering steps in unsolved patients and therefore represent a set of exons that are suspected to be deleted. Aiming to rank these exons, we plotted the ratio between MECG and MECI (figure 1B) in known IRD-causing genes. The analysis revealed 36 exons with a MECG/MECI ratio above 10 (see online supplementary table S1). Interestingly, some of these exons were consecutive within the same gene (eg, in NPHP4, MYO7A, CHM and RPGR) and are likely to represent genomic deletions including more than a single exon. In sample MOL0102-4, all CHM exons appear at high MECG/MECI values (figure 1B), indicating that the whole CHM gene is deleted in this patient. Aiming to examine the coverage of neighbouring genes on the X-chromosome, we performed a coverage analysis of the entire X-chromosome. The analysis revealed six additional genes that are suspected to be fully deleted in this sample (figure 1C).
Based on this analysis, we identified five predicted homozygous deletions and three predicted hemizygous deletions (see online supplementary table S1). Aiming to verify these deletions, we designed primers for the predicted deleted exons and verified six of the suspected deletions. The two non-verified deletions were suspected single-exon deletions showing the lowest MECG values (10.49 in TUB and 10.08 in CSPP1) therefore representing false-positive results. Aiming to identify the two borders of each of the six true deletions, we used the PCR-walking analysis (see Methods section) on genomic DNA samples.
Homozygous deletions
The analysis revealed three homozygous large deletions in the EYS, MYO7A and NPHP4 genes as detailed below.
EYS—we recruited for the study 2 affected and 10 unaffected individuals from a non-consanguineous Arab Muslim family with non-syndromic AR-RP (MOL0255). Although the parents did not report consanguinity, they were born at the same city and shared the same ethnic origin. We therefore performed homozygosity mapping on the two affected siblings and identified two homozygous regions of 8.5 and 6.6 Mb on chromosomes 6 and 14, respectively (figure 2A), the former includes EYS. In parallel, we excluded all known RP disease-causing mutations by using a genotyping array (at Asper ophthalmic). We then performed WES on the DNA sample of the index case and did not identify any potential disease-causing mutations. Exon deletion analysis revealed a MECG/MECI ratio of 24.95 in exon 29 of EYS (figure 1B), while the neighbouring exons were covered by a high number of reads. PCR walking analysis followed by direct Sanger sequencing revealed a large genomic deletion of 76 789 bp including exon 29 (figure 2B, D and table 1), which is expected to cause a frameshift, p.(Gln1977Cysfs*12). The 5′ breakpoint is located 41 bp upstream a LINE/L2 element (figure 2B, D). Interestingly, the 3′ breakpoint is located within a transposable element that belongs to the hAT-AC family.
Verified deleted exons as the cause of inherited retinal diseases
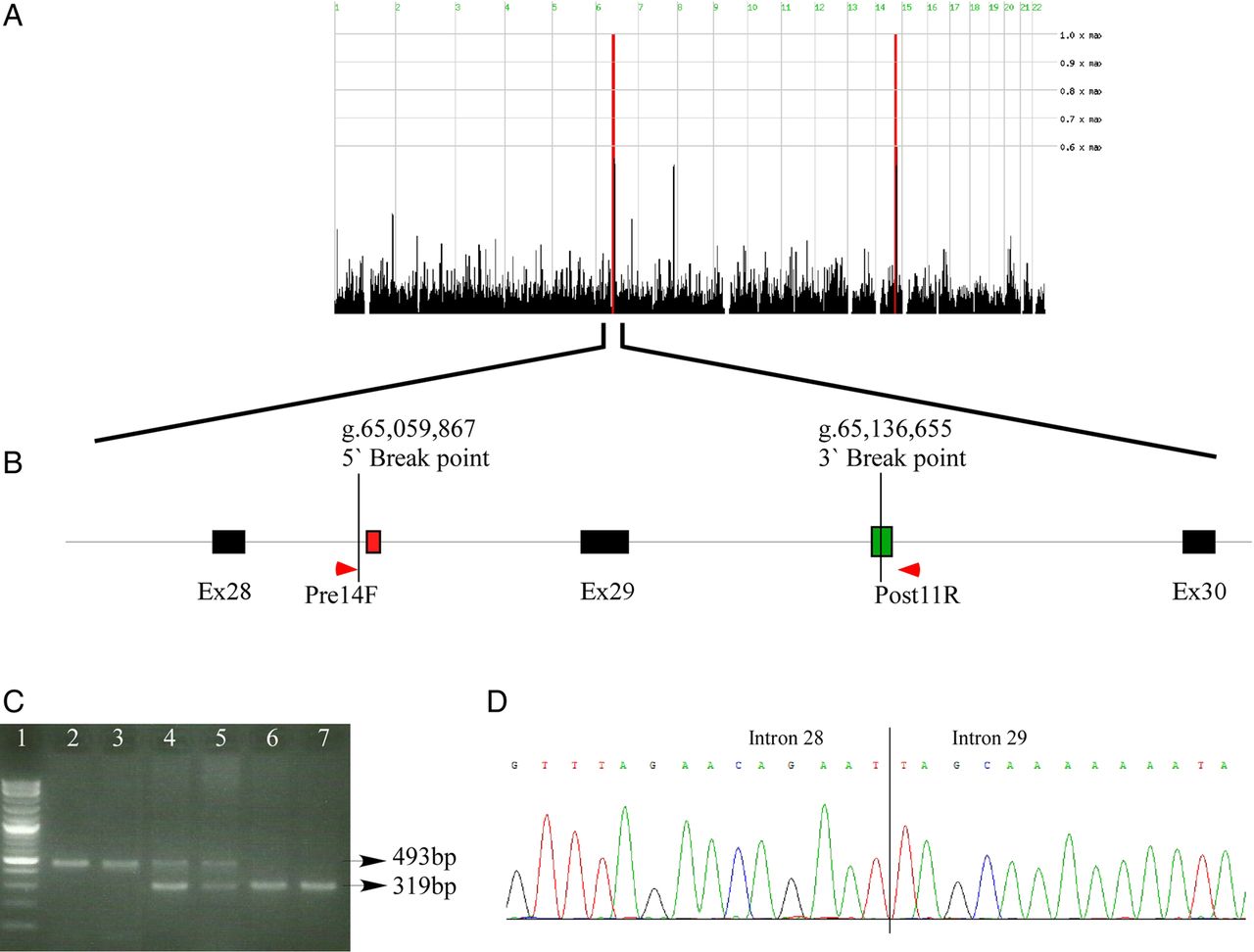
A homozygous genomic deletion in EYS. (A) Homozygosity mapping analysis of the two affected siblings in family MOL255. The two red areas represent large homozygous regions that are shared by the two siblings on chromosomes 6 and 14. (B) A schematic representation of part of the EYS gene showing the region of the genomic deletion. Black boxes represent coding exons, vertical lines depict the deletion breakpoints, red and green boxes represent LINE/L2 and hAT-AC transposon, respectively. Primer locations are shown as red arrows. (C) An agarose gel showing amplification through the deleted region. The lower band (319 bp) represents the wildtype (WT) allele using Ex29F and Ex29R primers and the upper band (493 bp) represents the mutated allele using Pre14F and Post11R primers (see online supplementary table S3). Lanes: 1—DNA ladder, 2—index case (homozygous), 3—affected sister (homozygous), 4—unaffected mother (heterozygous), 5—unaffected father (heterozygous), 6—unaffected sister (WT), 7—unaffected brother (WT). (D) A chromatogram showing the sequenced borders of the deleted area in introns 28 and 29.
MYO7A—WES coverage analysis of the index case of family MOL0467 (North African Jewish origin diagnosed with USH1—online supplementary table S4) revealed a high MECG/MECI ratio in three consecutive exons (including the terminal exon, figures 1B and 3A–C). A similar deletion was also suspected in another family (TB170) of the same origin by manual inspection of WES alignment. We were able to design primers producing a PCR product encompassing the deleted region (figure 3D) and identified the deletion borders within intron 46 and the 3′ untranslated region (3′UTR) of MYO7A (figure 3E and table 1) in both families (MOL0469 and TB170). The deletion size was determined as 1923 bp. A screen for this deletion in a set of eight additional USH index patients of the same origin did not reveal the mutation in either a heterozygous or a homozygous state. Sequence analysis of the deletion borders did not reveal any palindromic or repetitive elements in the deletion breakpoint area.

A genomic deletion in the MYO7A gene. (A and B) The genomic organisation of the MYO7A gene. (C) Whole exome sequencing (WES) alignment data (as viewed by integrative genomic viewer (IGV)) of the index case in MOL0467 show lack of specific reads in exons 47–49 (lower panel), compared with the adjacent exons as well as a control WES sample (upper panel). (D) Gel electrophoresis of PCR analysis using the Exon47F and Exon49R primers of homozygous patients (lanes 2 and 5), heterozygous family members (lanes 3 and 4) and a WT control (lane 6) verifying the deletion. DNA ladder is shown in lane 1. E Sequence analysis through the deleted area showing the deletion borders.
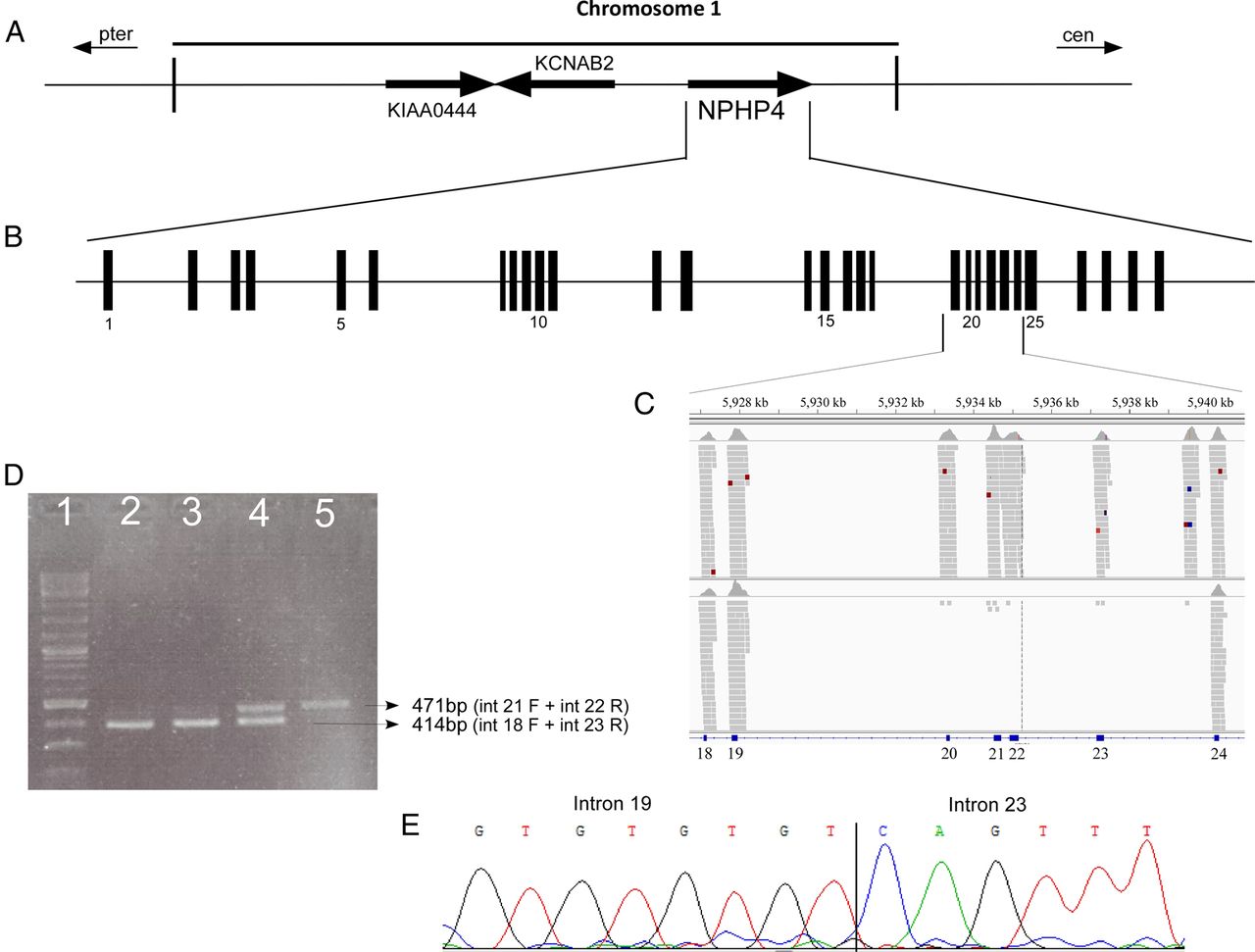
NPHP4—family MOL1268 of Arab Muslim origin includes two siblings affected by Senior–Loken syndrome due to a combined diagnosis of early-onset RP and renal failure, which resulted in kidney transplantation at the age of 35 years in both patients (see online supplementary table S4). No families of this origin were reported previously with this syndrome, and we therefore performed WES and identified a high MECG/MECI ratio (ranging from 60 to 529) in four consecutive NPHP4 exons (figures 1B and 4A–C). The deletion borders are found in introns 19 and 23 (figure 4C–E) and the deletion size is 7272 bp; IVS19+75 through IVS23+484. Sequence analysis of the deletion borders revealed that the 5′ break point occurred within a complex simple sequence repeat with (GT)4 upstream the breakpoint followed by (GT)3GCATGC(GT)3GCA(TG)7. No repeats were identified in the 3′ break point and no palindromic sequences were detected at both ends.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A genomic deletion in the NPHP4 gene. (A and B) The genomic organisation of the NPHP4 gene. (C) Whole exome sequencing (WES) alignment data (as viewed by IGV) of the index case in MOL1268 show lack of specific reads in exons 20–23 (lower panel) compared with the adjacent exons and to a control WES sample (upper panel). (D) PCR analysis of homozygous patients (lanes 2 and 3), a heterozygous family member (lane 4) and a WT control (lane 5) verifying the deletion. (E) Sequence analysis through the deleted area showing the deletion borders.
Hemizygous deletions
The analysis also revealed three hemizygous large deletions: two in the RPGR gene and one including seven consecutive XL genes. The two RPGR deletions were identified in two XLRP families (see online supplementary table S4) of Ashkenazi Jewish origin: a genomic deletion of 5786 bp including exons 2 through 5 (expected to cause an inframe deletion) in family MOL0054 and a deletion of 28 984 bp including exons 4 through 11 (also expected to cause an inframe deletion) in family TB162. Both mutations were not found in a set of 60 Ashkenazi Jewish patients with XLRP or isolate males with RP. The borders of the RPGR exons 2–5 deletion are IVS1-875 and IVS5+170 (see online supplementary figure S2). The 5′ deletion site does not contain any significant palindromic sequences but is located between a mammalian-wide interspersed repeat (MIR) sequence (ending 120 bp upstream the breakpoint) and an Alu sequence (starting 46 bp downstream the breakpoint). The 3′ breakpoint also does not contain any significant palindromic sequences. The deletion breakpoint, however, is within a simple sequence repeat of (TTTATGCTTA)4. The breakpoint occurred between the third and fourth nucleotide of the first repeat (see online supplementary figure S2). The deletion of RPGR exons 4–11 created an inframe deletion: p.A83_P471del.
Family MOL0102 (see online supplementary figure S1) includes three brothers who were initially diagnosed with XLRP but were negative for RP2 and RPGR mutations. WES analysis that was performed on the index case revealed 14 exons that belong to the CHM gene on X-chromosome showing high MECG/MECI ratios (figure 1B and online supplementary table S1), indicating a deletion of the whole CHM gene that is known to cause choroideremia when mutated. Aiming to define the deletion borders, we performed a careful analysis of WES data on the X-chromosome (figure 1C) that clearly showed deletion of seven consecutive genes on the X-chromosome (CHM, DACH2, KLHL4, CPXCR1, TGIF2LX, PABPC5 and PCDH11). A clinical re-evaluation of the index case at the age of 31 years revealed typical signs of choroideremia (see online supplementary table S4) including widespread chorioretinal atrophy extending from the periphery to the maculas (see online supplementary figure S3A). Clumps of pigment were also seen in the near periphery. FAF showed the typical picture of practically total chorioretinal atrophy except an irregular hyperfluorescent area in the centre of the macula (see online supplementary figure 3B). Besides CHM, none of the genes in the deleted region has been associated with a human disease. It should be noted, however, that the three siblings of the index case have been diagnosed with congenital hydronephrosis and were operated in early childhood. The index case does not suffer from any of these symptoms. We were not able to determine the deletion borders due to a large number of repetitive elements at the deletion borders.
Discussion
It is widely accepted that standard mutation analysis by Sanger sequencing of the coding regions and neighbouring intronic sequences usually results in the identification of about 80–90% of the mutations in monogenic diseases.24 ,25 The remaining mutations are considered as ‘deep intronic’ or large genomic rearrangements, mainly heterozygous ones. In heterogeneous diseases, such as RP, detection rate is much lower since the total number of genes that are associated with the disease is still unknown. In IRDs, mutation identification using WES or NGS-based targeted gene panels ranges from 40–60% of analysed samples.18–22 ,26 ,27 While WES cannot aid in the identification of deep intronic mutations, the obtained WES data can allow one, by using non-classical analysis approaches, to identify hemizygous, homozygous as well as heterozygous large exon deletions.
Here, we describe a new computational tool that can serve for efficient detection of homozygous and hemizygous deletions of at least a single exon by studying WES coverage data. Our analysis of 60 WES samples revealed the identification of six deletions (10%). This result emphasises the need to efficiently analyse WES data for all possible variants. To predict the impact of such deletions, we tabulated all disease-causing mutations (a total of 270 mutations) identified so far in our cohort of patients with IRDs. The majority of mutations are missense (119 mutations, 44.1%), followed by frameshift due to small insertions or deletions (60, 22.2%), nonsense (48, 17.8%), splice-site (28, 10.4%), large deletions of at least a single exon (11, 4.1%) and small inframe deletions (4, 1.5%). While most mutation types can be easily detected by Sanger or WES analysis at homozygous or heterozygous states, large deletions can often be overlooked. It is therefore reasonable to assume that our cohort contains such deletions that were not detected so far and that the fraction of large deletions (4.1% of all mutations) is an underestimate. Nonetheless, the more important fraction is that detected in WES samples (10%) and therefore each and every assessment of WES data must include an analysis of coverage data to detect such mutations. Alternatively, one should use whole genome sequencing analysis to detect genomic rearrangements relatively efficiently as reported recently.14
Recent studies used different analysis tools of WES data aiming to obtain some initial information regarding the possibility of CNV occurrence at multiple known loci.28 ,29 The analyses are based on coverage data of specific genomic regions, which need to be at a minimal quality to ensure reliability. A few WES-based approaches have been developed for CNV detection including ExomeCNV,30 CONIFER31 and CEQer.32 Although developed for detecting CNVs at a genomic view, these tools were reported to be applicable mainly in targeted analysis of a specific genomic locus and were reported to indeed yield reliable results.29 The most challenging task of these tools is to effectively filter out false-positive CNVs when analysing a large set of data with no previous knowledge of the deletion location. This is mainly critical for WES data with a relatively low base coverage of <100. Aiming to overcome this limitation, which is critical for heterogeneous diseases such as IRDs, we used a few filtering steps (including an efficient step, ie, based on the threshold of coverage found in WES data of females regarding genes that are located on the Y chromosome) resulting in an average reduction of suspected deletions to a value of 0.13% of the initial suspected exons (see online supplementary table S2). This might be the basis for an efficient genome-scale analysis that will not be limited to a specific genomic locus or a specific set of genes, but will enable one to search any gene that is covered by the high-throughput sequencing data.
One possible explanation for the mechanism of these deletions comes from the EYS deletion in Family MOL0255. Interestingly, the 3′ breakpoint is located within a transposon that belongs to family hAT-AC. A previous survey of hAT-AC in different genomes identified seven such elements in the human genome and the authors predicted that 70 such sequences are expected.33 These sequences may cause mutations by ‘jumping’ into new genomic sites in the genome.
In summary, we show here that WES data can be analysed efficiently for detecting homozygous large deletions that comprise about 10% of our WES samples.
Acknowledgments
The authors thank all patients and family members for their participation in this study.
References
Footnotes
Contributors SK, MH, AH, AB, SM, AA-D, FAT, LM-M and TB-Y performed experimental work, data analysis and revised the manuscript. SK, SL and EB performed clinical work and revised the manuscript. DS and EB designed and directed the project. DS, SK, MH and EB wrote the manuscript.
Funding This work was supported by the Foundation Fighting Blindness USA grant number BR-GE-0214-0639 (to DS, EB and TB-Y), by the Israel Science Foundation grant number 2154/15 (to SK), by the Chief Scientist Office of the Israeli Ministry of Health and the Lirot association grant number 300009177 (to SK) and the Yedidut Research grant (to EB).
Competing interests None declared.
Ethics approval The research was approved by the institutional review board (IRB) at the Hadassah Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.