Article Text

Abstract
Background Alport syndrome is a clinically heterogeneous, progressive nephropathy caused by mutations in collagen IV genes, namely COL4A3 and COL4A4 on chromosome 2 and COL4A5 on chromosome X. The wide phenotypic variability and the presence of incomplete penetrance suggest that a simple Mendelian model cannot completely explain the genetic control of this disease. Therefore, we explored the possibility that Alport syndrome is under digenic control.
Methods Using massively parallel sequencing, we identified 11 patients who had pathogenic mutations in two collagen IV genes. For each proband, we ascertained the presence of the same mutations in up to 12 members of the extended family for a total of 56 persons studied.
Results Overall, 23 mutations were found. Individuals with two pathogenic mutations in different genes had a mean age of renal function deterioration intermediate with respect to the autosomal-dominant form and the autosomal-recessive one, in line with molecule stoichiometry of the disruption of the type IV collagen triple helix.
Conclusions Segregation analysis indicated three possible digenic segregation models: (i) autosomal inheritance with mutations on different chromosomes, resembling recessive inheritance (five families); (ii) autosomal inheritance with mutations on the same chromosome resembling dominant inheritance (two families) and (iii) unlinked autosomal and X-linked inheritance having a peculiar segregation (four families). This pedigree analysis provides evidence for digenic inheritance of Alport syndrome. Clinical geneticists and nephrologists should be aware of this possibility in order to more accurately assess inheritance probabilities, predict prognosis and identify other family members at risk.
- Complex traits
- Diagnostics tests
- Genetic screening/counselling
- Getting Research into Practice
- Molecular genetics
Statistics from Altmetric.com
- Complex traits
- Diagnostics tests
- Genetic screening/counselling
- Getting Research into Practice
- Molecular genetics
Introduction
Alport syndrome is an inherited nephropathy characterised by haematuria, proteinuria, progressive renal failure and ultrastructural lesions of the glomerular basement membrane (GBM), often associated with sensorineural deafness and ocular lesions.1 ,2 The primary defect resides in one of the α chains of type IV collagen produced by podocytes; the α chains assemble into a heterotrimeric triple helix (α3, α4 and α5) to create the three-dimensional network of the basement membrane.3 The α3 and α4 chains are encoded by COL4A3 and COL4A4 genes, located head-to-head on chromosome 2,4 while the α5 chain is encoded by COL4A5 on chromosome X.5 All three main models of Mendelian inheritance have been demonstrated in Alport syndrome: X-linked semidominant, autosomal recessive and autosomal dominant.6–9 X-linked semidominant inheritance is associated with mutations in COL4A5, while autosomal-dominant and autosomal-recessive inheritance patterns are associated with one or two mutations, respectively, in either COL4A3 or COL4A4.4 ,10
Mutant alleles of all three primary loci demonstrate variable expressivity. In particular, heterozygous COL4A5 women may be asymptomatic or have symptoms that range from haematuria alone to severe nephropathy leading to end-stage renal disease (ESRD).11 Similarly, heterozygous carriers of COL4A3 or COL4A4 mutations, irrespective of gender, may be asymptomatic, may have haematuria (carriers of recessive disease) or may progress to ESRD, although at a later age (apparently dominant form).12 ,13 A genotype–phenotype correlation between COL4A5 mutations and both the rate of progression to ESRD and the course of hearing loss and ocular lesions has been established in X-linked Alport syndrome, but, even within a single family, there is variability in disease severity.11 ,14 Part of this variability may be due to a modifier effect of functional polymorphisms in the three collagen chains (of which COL4A3 and COL4A4 are particularly rich) or in other structural proteins of GBM, such as podocin.15 Clarification of this relevant clinical variability warrants further investigation.
The wide phenotypic variability of patients with Alport syndrome and the presence of incomplete penetrance suggest that a simple Mendelian model is inadequate to explain the genetic control of this disease. An alternative genetic model that may apply to Alport syndrome is digenic inheritance. As explained by Schäffer in 2013, “inheritance is digenic when the variant genotypes at two loci explain the phenotypes of some patients and their unaffected (or more mildly affected) relatives more clearly than the genotypes at one locus alone”.16 Digenic inheritance has been demonstrated in numerous diseases such as deafness,17–21 retinitis pigmentosa22 and left ventricular non-compaction.23 During the past few years, the knowledge of two-loci diseases has improved due to extensive use of comparative genomic hybridisation and microarray technology (array CGH). In particular, it has recently been shown, in patients with various neuropsychiatric diseases, that an enrichment in copy number variants (CNVs) correlates with a more severe phenotype, that is, a specific microdeletion both predisposes to neuropsychiatric phenotypes as a single event and exacerbates neurodevelopmental phenotypes in association with other deletions or duplications.24 ,25
Research into the genetic causes of disease has been facilitated by the recent development of massively parallel sequencing techniques. This technology permits the sequencing of many genes simultaneously as a routine procedure9 ,12 ,26 and should accelerate the discovery and characterisation of disorders governed by digenic inheritance.16 In the past, when Sanger sequencing was the dominant method, it was common to stop analysis when a single pathogenic mutation was identified; this approach prevented the ascertainment of individuals with mutations in more than one collagen gene. Since 2011, we have been using massively parallel sequencing to evaluate the three collagen IV genes in patients with Alport syndrome, and this approach permitted us to identify 11 people who have mutations in two genes (molecular data for five patients have already been published).26 In this study, we examined the mutation pattern and clinical characteristics of these 11 patients and members of their extended families to explore the possibility that Alport syndrome is under digenic control.
Materials and methods
Patients and families
Genetic counselling was performed in six European institutes (University of Siena, Italy; Hôpital Necker—Enfants Malades, Paris, France; Guy's Hospital, London, England; Newcastle upon Tyne Hospitals NHS Foundation Trust, UK; Maastricht University Medical Centre, The Netherlands; and Université Catholique de Louvain, Belgium) where patients with Alport syndrome were selected for mutation screening in the COL4A3, COL4A4 and COL4A5 genes according to clinical criteria. These included a positive family history, a lamellated GBM, high tone sensorineural hearing loss, and lenticonus and macular flecks on ophthalmoscopy.27 Eleven unrelated persons were found to have pathogenic mutations in more than one collagen gene. These patients (four men and seven women, 3–55 years old) and members of their extended families were recruited for the present study, allowing us to construct 11 pedigrees. From all persons recruited, we collected clinical data regarding family status (proband or family relation), gender and age at inclusion (or death), kidney function (haematuria, proteinuria, chronic renal failure (CRF) or ESRD), hearing loss and ocular lesions; for all comorbidities, we recorded age at diagnosis and eventual treatment. Moreover, we obtained a sample of peripheral blood in EDTA tubes when possible.
Genomic DNA, amplification and sequencing strategy
Blood was stored frozen prior to the extraction of genomic DNA using QIAamp DNA Blood Kits (Qiagen, Hilden, Germany); this work was done at each institute for the families recruited there. In Siena, genomic DNA was supplied directly from the institutional biobank.
Genomic DNA from probands was assessed for mutations in COL4A3, COL4A4 and COL4A5 by locus-specific amplification followed by massively parallel sequencing. Each participating institute amplified and sequenced the DNA for the probands it recruited. Briefly, the ALPORT MASTR kit (Multiplicom, Niel, Belgium) was used to amplify 149 amplicons (representing 150 coding exons) of the three genes in a four-tube multiplex PCR reaction starting with about 4×50 ng genomic DNA. Amplification products were sequenced using either a GS Junior System (454 Life Sciences, Roche) or an Ion Personal Genome Machine (PGM; Life Technologies), as described below. Mutations of Probands 3, 4, 5, 7 and 9 have already been reported in Morinière et al.26 Genomic DNA from family members was analysed by Sanger sequencing to determine whether these persons had the same mutations as the probands.
GS Junior 454 sequencing
Our strategy for sequencing the COL4A3, COL4A4 and COL4A5 genes on a GS Junior system has been reported.9 Briefly, amplification products were diluted and then reamplified with primers containing, at the 5′ end, a multiplex identifier sequence that barcodes the samples. These PCR products for each proband were pooled in predefined proportions according to the ALPORT MASTR protocol (Multiplicom). These libraries were purified using Agencourt AMPure XP system (Beckman Coulter) and quantified using Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies), following the protocol of 454 Life Sciences (http://454.com/downloads/my454/documentation/gs-junior-plus/454SeqSys_Amplicon-Library-Prep-MM_Apr2014.pdf).
For GS Junior sequencing, libraries were diluted to a concentration of 1×107 PCR fragment molecules/µL, annealed to carrier beads (SeqCap EZ Pure Capture Bead Kit, Roche NimbleGen) and clonally amplified by emulsion PCR (emPCR) according to the manufacturer's protocol (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJunioremPCRAmplificationMethodManualLib-A_March2012.pdf). After amplification, the beads carrying single-stranded DNA templates were enriched, counted and deposited into the PicoTiterPlate for sequencing (http://454.com/downloads/my454/documentation/gs-junior/method-manuals/GSJuniorSequencingManual_Jan2013.pdf). Sequence reads were analysed using GS Amplicon Variant Analyzer, V.2.9.
Ion PGM sequencing
The strategy for sequencing the three collagen genes on an Ion PGM has recently been reported.26 Briefly, amplification products were diluted, reamplified using the universal primers included in ALPORT MASTR kit and pooled in predefined proportions according to the ALPORT MASTR protocol. This pool was used to prepare a barcoded library compatible with the Ion PGM according to the protocol of Life Technologies (http://ioncommunity.lifetechnologies.com/community/login.jspa?referer=http://ioncommunity.lifetechnologies.com/community/protocols-home).26 Libraries were purified using Agencourt AMPure XP system and quantified using Qubit dsDNA HS Assay Kit (Life Technologies).
For Ion PGM sequencing, PCR fragments were diluted to 100 pM, annealed to carrier spheres (Ion Sphere Particles) and clonally amplified by emPCR using the Ion PGM Template OT2 200 kit (Life Technologies). Spheres carrying single-stranded DNA templates were transferred to Ion 314 chips for sequencing using the Ion PGM Sequencing 200 Kit V.2. Data were processed using Torrent Suite software V.4.0, while postrun analysis was conducted using Torrent Variant Caller plug-in (V.4.0-r72895).
In silico analyses
Pathogenicity of variants was determined in accordance with the Clinical Molecular Genetics Society best practice guidelines.28 Pathogenicity was ascertained if the following criteria were met: non-polymorphic missense mutations or in-frame deletions involving key amino acids, such as glycine in the collagen Gly-X-Y triple helical domain, splice-site mutations and truncating mutations. Pathogenicity of non-synonymous variations other than Gly substitutions was predicted using Alamut software V.2.3 (Interactive Biosoftware, Rouen, France), which includes the tools Align GVGD, SIFT, MutationTaster, PolyPhen-2, SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer, Human Splicing Finder and ESE tools.
To determine whether the identified sequence variants were novel or had been previously reported, we searched in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php),29 the Leiden Open Variation Database V.2.0 Build 35 (https://grenada.lumc.nl/LOVD2/COL4A/variants.php?select_db=COL4A3&action=search_all&search_MutCol=%3E) and the ALPORT (COL4A5) database (http://www.arup.utah.edu/database/ALPORT/ALPORT_display.php?sort=2#alport; last update: October 2013).30
Variants were described according to the COL4A3 reference sequence LRG_230 (NM_000091.4), COL4A4 reference sequence LRG_231 (NM_000092.4) and COL4A5 reference sequence LRG_232 (NM_000495), where nucleotide number 1 corresponds to the first base of the translation initiation codon, and using the nomenclature recommended by the Human Genome Variation Society.31
Sanger sequencing and segregation analysis of pedigrees
Pathogenic variants identified in probands were confirmed by direct Sanger sequencing. Sanger sequencing was also used to determine whether the pathogenic variants were present in family members for whom genomic DNA was available. Briefly, genomic DNA was amplified using the primers and PCR conditions described for amplicon library preparation in Artuso et al.12 Sequencing was performed on an ABI Prism 310 genetic analyser (PE Applied Biosystems) and data were analysed with Sequencher software V.4.9 (Gene Codes, Ann Arbor, USA). Genotypes of pedigrees were examined to determine whether the COL4A3 and COL4A4 mutations on chromosome 2 were inherited independently on different homologous chromosomes or not, like in trans or in cis, and to assess genotype–phenotype correlation. In the present paper, families with similar inheritance patterns were reported with consecutive numbers.
Results
The study began with 11 patients with Alport syndrome in whom massively parallel sequencing identified pathogenic mutations in two of the three collagen IV genes examined. In seven patients (probands 1–7), there was a combination of mutations in COL4A3 and COL4A4, whereas in four (probands 8–11) one or two mutations in COL4A4 associated with a mutation in COL4A5. In no case were there simultaneous COL4A3 and COL4A5 mutations. Altogether, 23 unique mutations were found, including 7 in COL4A3, 12 in COL4A4 and 4 in COL4A5 (table 1). The mutations involved all domains of the collagen molecules, although the majority of missense mutations (11 of 13) affected the triple-helical collagenous domain and 11 missense mutations substituted a critical glycine residue in this domain. Overall, 13 mutations have been previously reported (among missense mutations six are already listed in dbSNP and four26 are pending assignment of a dbSNP reference ID) and 10 are novel.
Molecular features and predicted pathogenicity of 23 mutations in collagen IV genes, found in 11 patients with Alport syndrome
Between 1 and 12 family members were recruited for each proband, for an average of four members per family (table 2). Therefore, the study considered a total of 56 persons (27 men and 28 women; gender missing for one person) from 5 to 80 years of age (exact age missing for 11 persons). Seven individuals were dead at the time of study. A wide range of kidney functionality was observed in the study population, ranging from normal (in seven persons), to haematuria, proteinuria, CRF (in six persons) and ESRD (in 12 subjects leading to death in four cases). Hearing loss was recorded in 8 of 44 persons for whom hearing test results were available, and ocular lesions were noted in 2 of 6 persons for whom ophthalmological data were available.
Clinical characteristics and collagen gene mutations in 11 patients with Alport syndrome and their family members
Genomic DNA was available for 34 family members (all alive at the time of study). Sanger sequencing of this DNA revealed which family members had the same mutations as the probands (including which were heterozygous or hemizygous for only one mutation or compound heterozygous in only one gene), permitting us to explore the relationship between mutations and disease severity as well as to investigate the form of genetic transmission. Individuals with two mutations tended to be more severely affected than those with one mutation. The few cases of hearing loss and ocular lesions were observed only in persons with mutations in two genes, and, among the five persons with ESRD and sequencing data, the age at onset was lower in the two cases with two genes affected (25 and 44 years) than in those with one gene affected (family 4).
Family 1 was identified first (figure 1). In the proband (II:2) (figure 1A), massively parallel sequencing (figure 1B) revealed the presence of a COL4A4 glycine substitution inherited from the ascertained asymptomatic father and a splice site mutation in the COL4A3 gene inherited from the haematuric mother. The disruption of the terminal part of the triple helix in the α3 chain and the kink formed by the presence of the bigger glutamic acid instead of the flexible, small glycine can be assumed to prevent the correct formation of the triple helix, which assembles from the C-terminal tail (figure 1C). Although the proband was very young (7 years), the presence of recurrent episodes of macroscopic haematuria may indicate a poor prognosis.
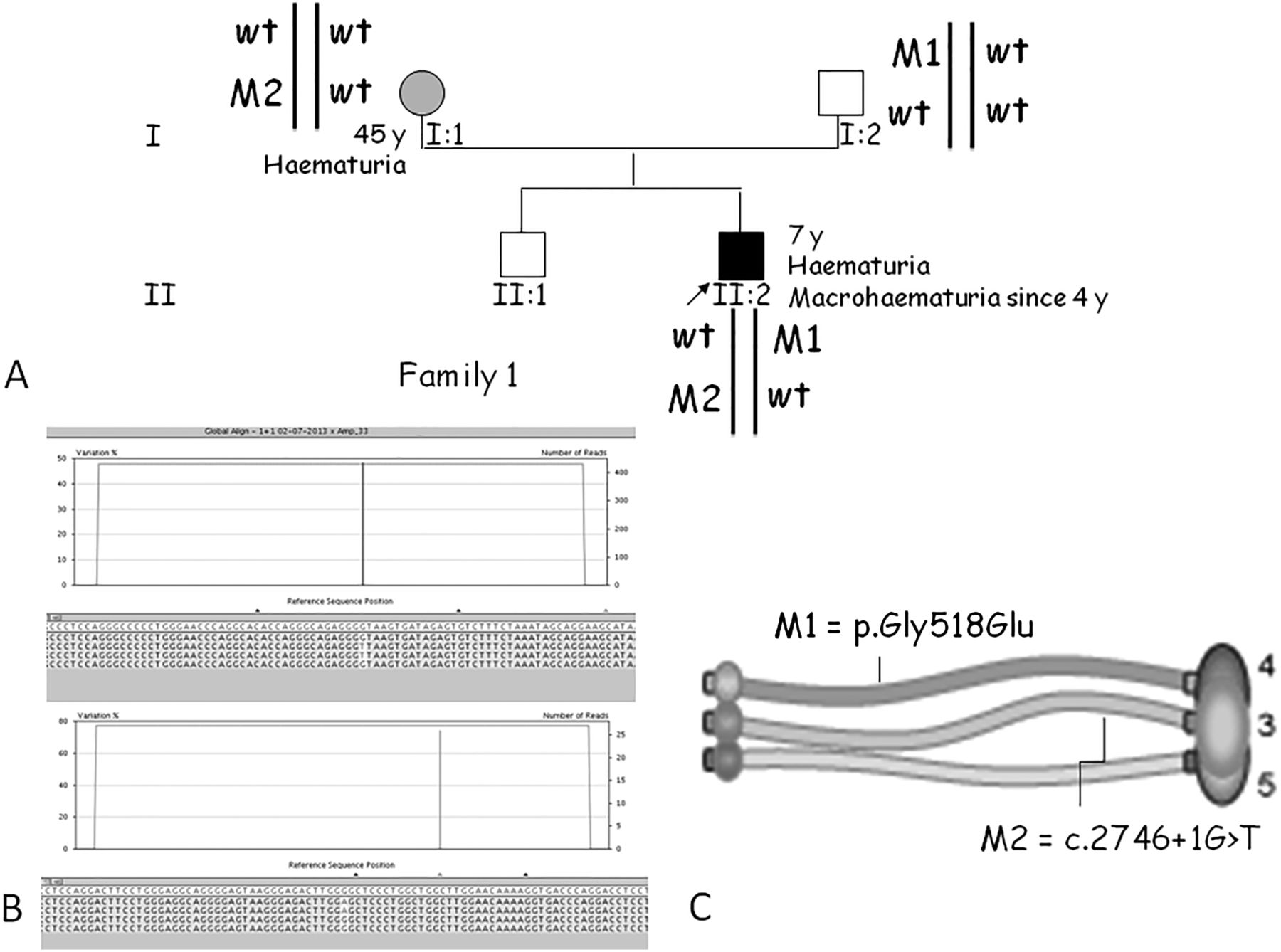
Molecular and segregation analysis of family 1. (A) Pedigree: the mutated alleles are in trans and the mode of inheritance is digenic autosomal. wt, wild-type allele. (B) Screenshots from the GS Amplicon Variant Analyzer software showing the position of the COL4A4 missense mutation c.1553G>A (p.Gly518Glu; M1) and the COL4A3 splice site variant c.2746+1G>T (M2). The upper histograms indicate the percentage of variation. In the lower panels, reads from different directions are displayed and the mutated base is highlighted. (C) Locations of these mutations on the a3-a4-a5 triple helix of collagen.
Digenic autosomal inheritance with mutations on different homologous chromosomes
In five of the seven families with COL4A3/COL4A4 mutation combinations, the two mutations were inherited independently, like in trans (families 1–5; figures 1 and 2). In these families, individuals with two heterozygous mutations had more severe phenotypes than those with a single heterozygous mutation. Family members having only one mutation in a collagen gene were asymptomatic (father in family 1) or had haematuria (mothers in families 1, 2 and 4; father in family 2; niece in family 3) or intermittent haematuria (daughters in family 3).
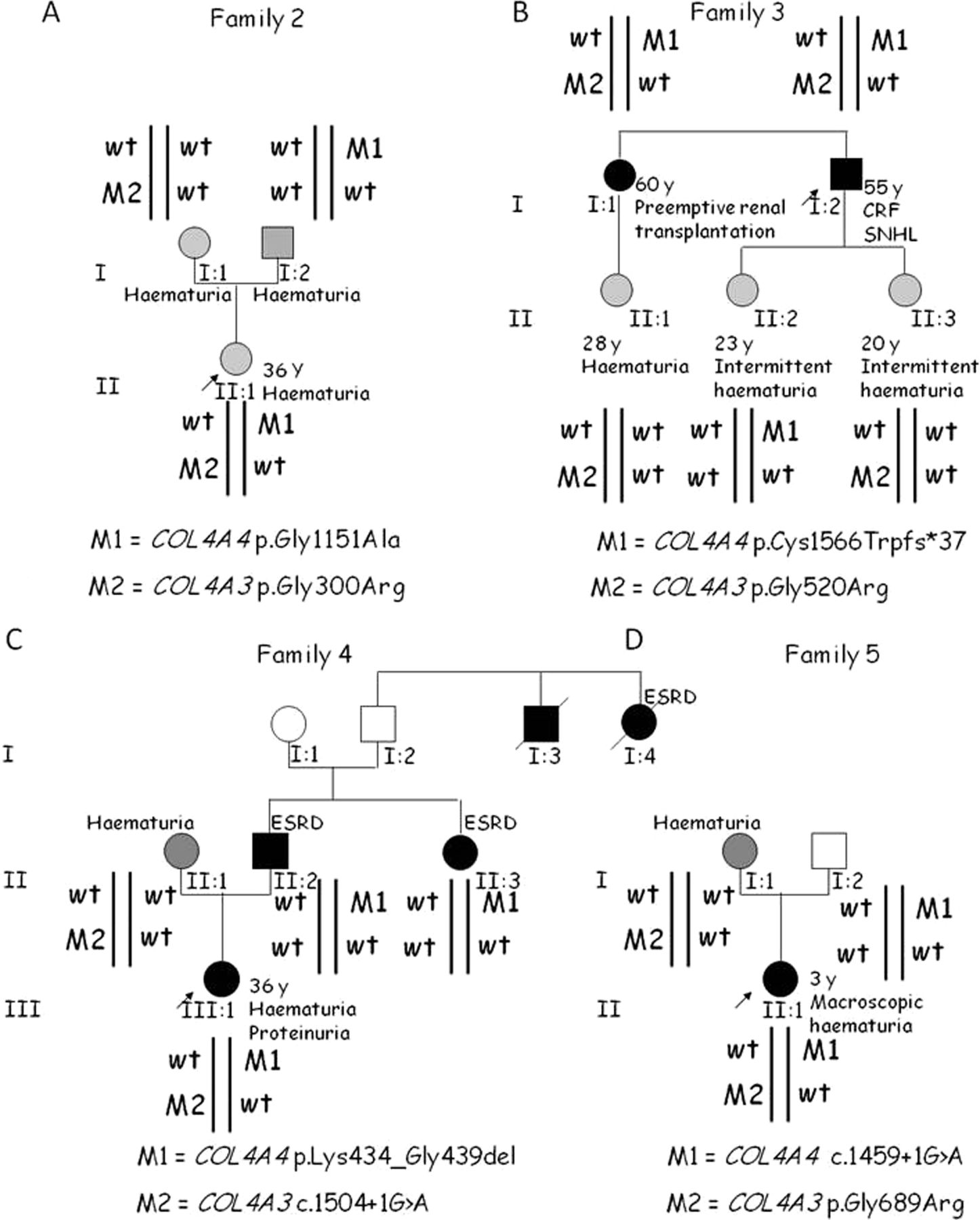
Pedigrees of four other families having digenic autosomal inheritance with mutations in COL4A3 and COL4A4 in trans. (A) Family 2. (B) Family 3. (C) Family 4. (D) Family 5. CRF, chronic renal failure; SNHL, sensorineural hearing loss; ESRD, end-stage renal disease.
In the digenic model with mutations on different chromosomes, the mode of inheritance resembles a recessive model, in the sense that the probability of having another child with the same genotype is 25%. The main difference is that the classic recessive model is determined by the combination of two alleles mutated at the same locus, while the digenic model is attributable to two different alleles mutated at two different loci. However, individuals with digenic disease have an intermediate phenotype between the autosomal-dominant and the autosomal-recessive forms with a severity of renal involvement, estimated according to the mean age at onset of ESRD, that is intermediate between those of the classic forms.
Digenic autosomal inheritance with mutations on the same homologous chromosome
In the other two families with COL4A3/COL4A4 mutation combinations, the two mutations were inherited together on the same chromosome, like in cis (figure 3). In families 6 and 7, the inheritance pattern resembles an autosomal-dominant mode: the probability of having another child with the same genotype is 50%, but the phenotype is more severe than expected for the classic autosomal-dominant form. In fact, subject I:2 of family 6 and subjects II:1 and I:2 of family 7 had CRF at an early age, with two of them progressing towards ESRD at 40 years of age, which is unexpected for the autosomal-dominant form.32

Families with digenic autosomal inheritance with mutations in COL4A3 and COL4A4 in cis. (A) Family 6. (B) Family 7. CRF, chronic renal failure; SNHL, sensorineural hearing loss; ESRD, end-stage renal disease.
Digenic unlinked autosomal/X-linked inheritance
In families 8–11, there was a combination of a mutation in the autosomal COL4A4 gene and in the X-linked gene COL4A5 (figures 4 and 5). In these families, double heterozygotes also have more severe phenotypes than expected for individuals with a COL4A4 heterozygous mutation or for COL4A5 carrier women.11 ,32 In fact, the female I:1 in family 8 had ESRD at the age of 44 years, earlier than expected had she had just one mutation in either COL4A4 or COL4A5.

Families with mutations in COL4A4 and COL4A5, with digenic unlinked autosomal and X-linked inheritance. (A) Family 8. (B) Family 9. (C) Family 10. SNHL, sensorineural hearing loss; ESRD, end-stage renal disease.

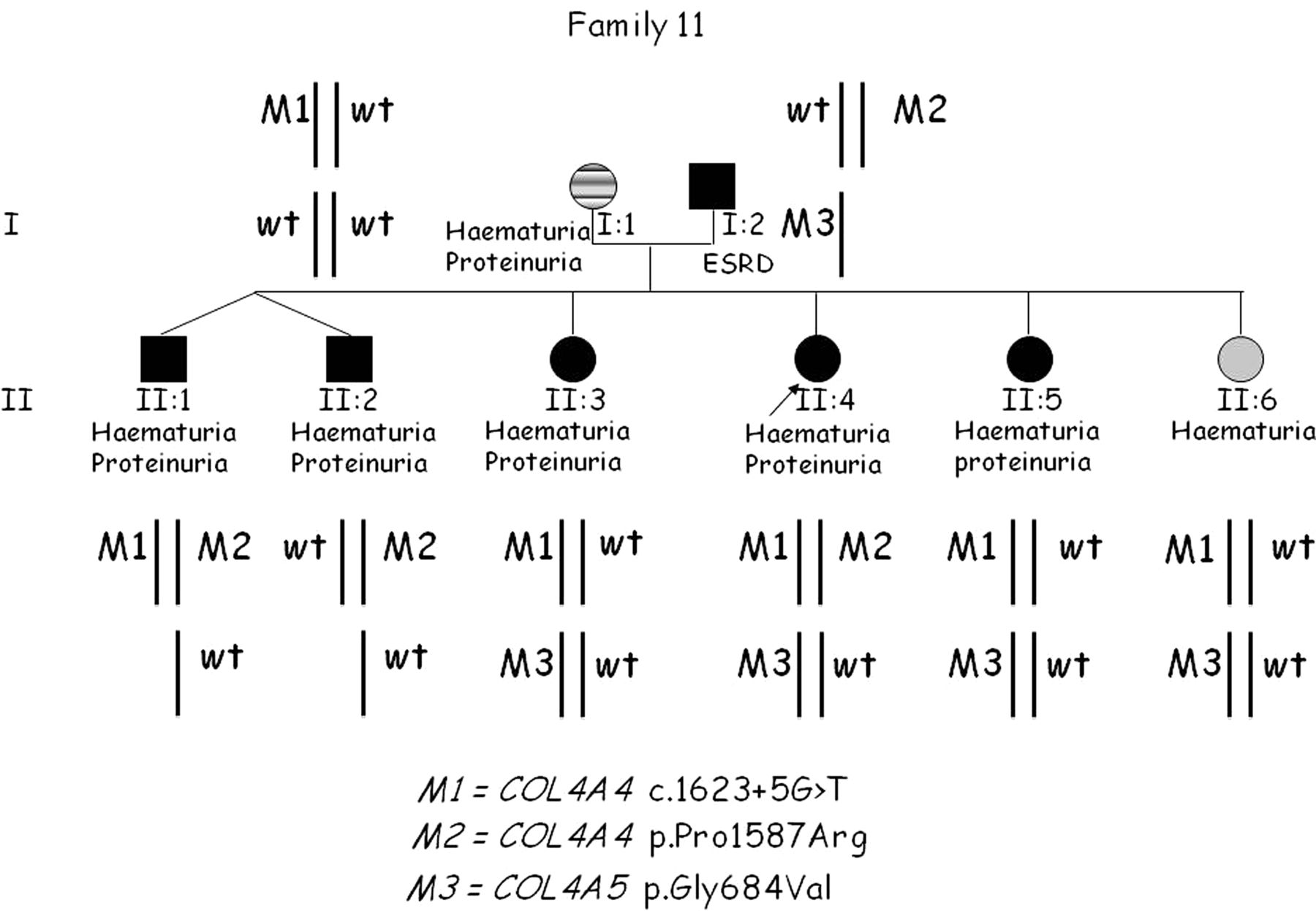
Pedigree of family 11 with a triallelic form of digenic inheritance, with mutations in COL4A4 and COL4A5. ESRD, end-stage renal disease.
In family 11, in addition to the mutation in COL4A5, two mutations in COL4A4 were found, resembling a triallelic inheritance (figure 5). The proband (II:4) presented with intermittent haematuria and proteinuria, and a first genetic testing by Sanger sequencing revealed an autosomal-recessive form of Alport syndrome due to compound heterozygosity at the COL4A4 gene. The first male sib (II:1) had both COL4A4 mutations, while three other sibs (II:2, II:3 and II:5) with a similar degree of the disease were carriers, as was the last sib who had isolated haematuria. At clinical re-evaluation, the father was found to have ESRD, so the proband was retested using massively parallel sequencing techniques. An additional pathogenic mutation resulting in a Gly substitution at codon 684 in COL4A5 was identified. Not surprisingly, all sisters were carriers. We suggest that the concomitant mutations in two different genes may be associated with a more severe clinical picture, even if in this family a follow-up is needed.
Discussion
The present study provides evidence that digenic inheritance can occur in Alport syndrome in addition to classic Mendelian inheritance. Using massively parallel sequencing, we investigated 11 families with variable degrees of clinical severity among their members who also varied in the genotype of two loci of two collagen type IV genes. In the reported pedigrees, the ‘two-locus model’ explains the variable expressivity of the disease within the same family better than simple Mendelian inheritance: the different genotypes at two loci, roughly equal in importance, can explain the differences in age at onset of renal failure and in the severity of the symptoms. This discovery has implications for genetic counselling especially for risk assessment of patients’ relatives because an erroneous definition of the inheritance model may result in incomplete cascade of testing relatives with consequent erroneous risk estimations.
All missense mutations, except two, affected glycine residues and are in the collagenous domain. Glycine is a small amino acid essential for making the protein flexible and allowing the coiling of the triple helix, building the final shape of the collagenous domain. The other two missense mutations were found in the main non-collagenous domain (NC1) relevant for the protein's self-assembly and formation of the irregular polygonal network. One such mutation, c.4760C>G in COL4A4, affects an evolutionarily conserved codon.33 A different mutation having the same effect on the protein's sequence, p.(Pro1587Arg), was recently reported in another patient with Alport syndrome.34 The other missense mutation, c.4994G>A (p.Cys1665Tyr) in COL4A3, eliminates a cysteine. Cysteine residues are key amino acids in the non-collagenous domains because their disulfide bridges are important for the globular structure. These observations strengthen the pathogenic classification of these changes.
In our cohort, we identified seven families with associated mutations in COL4A3 and COL4A4 genes and four families with associated mutations in COL4A4 and COL4A5. We did not find kindreds with digenic inheritance attributable to mutations in COL4A3 and COL4A5. This is likely due to the small size of our cohort; however, we cannot exclude a possible biological mechanism. Present knowledge of basement membranes is based on the 1:1:1 model. Each α chain (α3, α4 and α5) interacts equally with the other two, concurring to form a triple helix. Therefore, there is no molecular explanation for missing a COL4A3/COL4A5 combination. It is likewise unlikely that this combination gives rise to an unrecognisable phenotype.
In our cohort, double heterozygotes reached ESRD at the age of 40 years (subject I:2 of family 6 and I:2 of family 7) or 44 years (subject I:1 of family 8). It is interesting to note that this is later than the mean age expected in the autosomal-recessive form (31 years) but earlier than expected in the autosomal-dominant form (56 years).9 ,32 This fits well with the stoichiometry of the molecules of the triple helix (figure 6). In double heterozygotes, about 75% of triple helix molecules are expected to be defective, which is >50% in heterozygotes and <100% in homozygotes or hemizygotes.
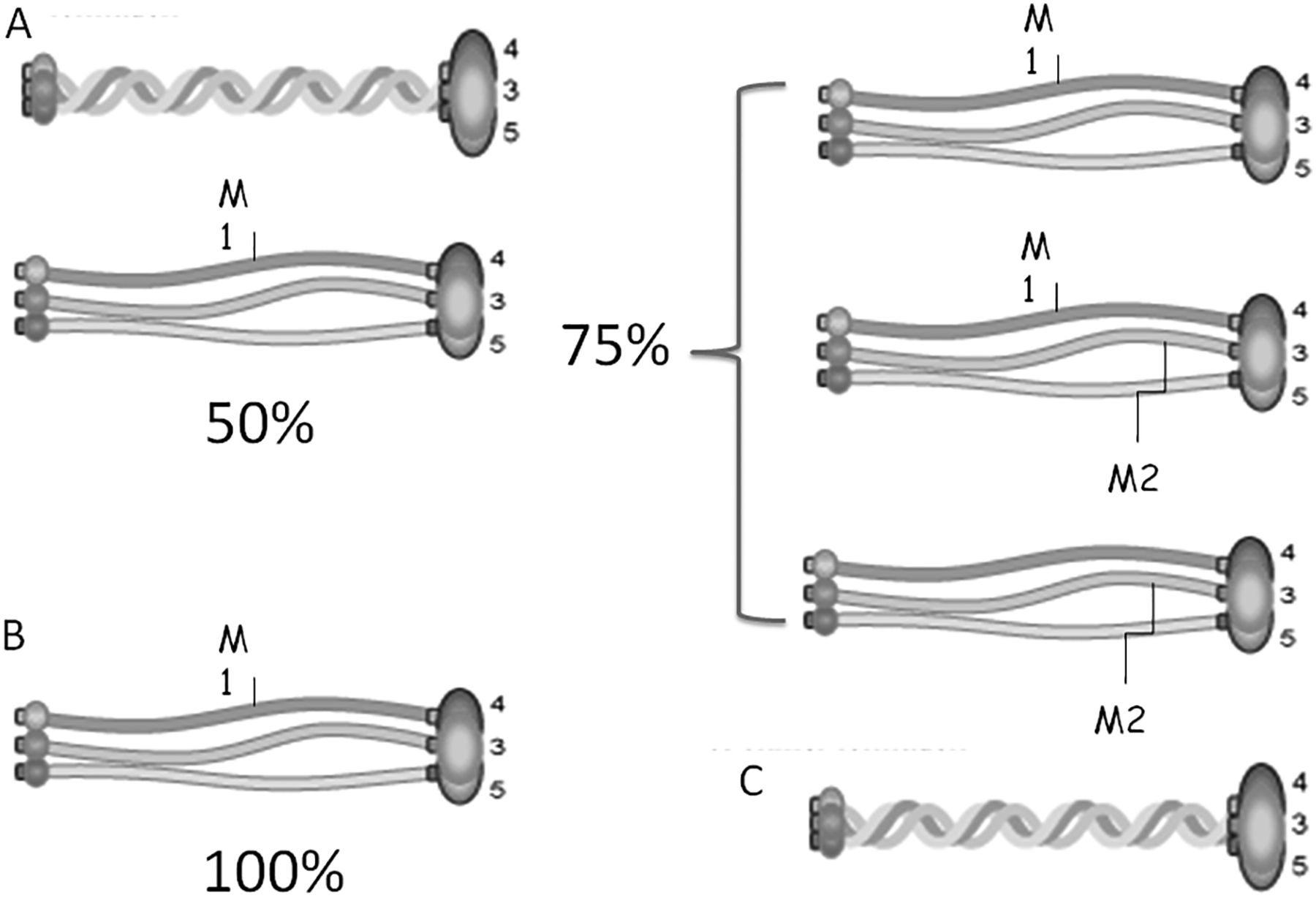
Triple helix combinations of defective α chains. (A) In heterozygotes, about 50% of triple helix molecules are expected to be defective. (B) In homozygotes or hemizygotes, 100% of triple helix molecules are expected to be defective. (C) In double heterozygotes, about 75% of triple helix molecules are expected to be defective.
Pedigree 11 may represent an example of the triallelic form of digenic inheritance. This kind of inheritance has been previously proved in Bardet–Biedl syndrome and other diseases.35 ,36 Triallelic inheritance is defined when any combination of three deleterious alleles at two loci, but not three heterozygous mutations at three loci, is sufficient to cause the disease. In the case of family 11, subject II:4 has two mutated alleles at the COL4A4 locus (M1, M2) in addition to one mutated allele at the COL4A5 locus (M3). In this family, an accurate follow-up of clinical progression may enhance our understanding of how the combination of different mutated alleles contributes to the developing phenotype. While the pathogenicity of the splice site mutation in COL4A4 (M1) and of the Gly substitution in the collagen domain of COL4A5 (M3) is certain, one could question the pathogenicity of the COL4A4 Pro to Arg substitution in the NC1 domain (M2; rs190148408). Two of three prediction tools scored this variant as pathogenic, and its reported frequency in the general population is 0.3%. Segregation analysis of family 11 is suggestive of a role of this allele in worsening the phenotype. Undoubtedly more data are necessary to exactly define the role of this mutation before we can conclude that the pedigree is an example of triallelic inheritance.
Digenic inheritance has already been reported in other renal pathologies besides Alport syndrome. For example, mutations in NPHS1 and NPHS2, two genes that encode the glomerular proteins nephrin and podocin, respectively, cause two distinct, severe forms of nephropathy. In three patients with a mutation in each gene (one homozygous and one heterozygous mutation), the combined mutations modified the disease phenotype from congenital nephrotic syndrome to congenital focal segmental glomerulosclerosis.37 In autosomal-dominant polycystic kidney disease, two patients with heterozygous mutations in both PKD1 and PKD2 had more severe disease than did patients with mutations in just one gene.38 In Bartter's syndrome, a spectrum of nephropathies due to impaired functioning of ion channels or transporters, two cases have been reported in which a severe form of the disease (associated with deafness) was attributed to mutations in CLCNKA and CLCNKB, adjacent genes encoding chloride channels.39 ,40 These two channels are expressed in the epithelial cells of both the kidney and the ear. Finally, at least one other collagen disorder has been proposed to have digenic inheritance. In Ullrich congenital muscular dystrophy, a disease caused by mutations in any of the genes encoding collagen type VI, namely COL6A1, COL6A2 and COL6A3, digenic inheritance has been reported for one patient who was compound heterozygous for mutations in COL6A1 and COL6A2.41 Together, these cases illustrate how, in complex syndromes with variable severity of symptoms, digenic inheritance may be playing a role in the clinical expression and course of the disease. In summary, in this paper we provide evidence for a digenic inheritance model for Alport syndrome and illustrate three possible segregation models: (i) autosomal inheritance with mutations on different homologous chromosomes (in trans) resembling the recurrence risk of a recessive disease; (ii) autosomal inheritance with mutations on the same chromosome (in cis) resembling the recurrence risk of a dominant disease and (iii) unlinked autosomal and X-linked inheritance having its own distinctive segregation. While the first case (in trans) represents a novelty with purely scientific interest, the other two have important implications in genetic counselling.
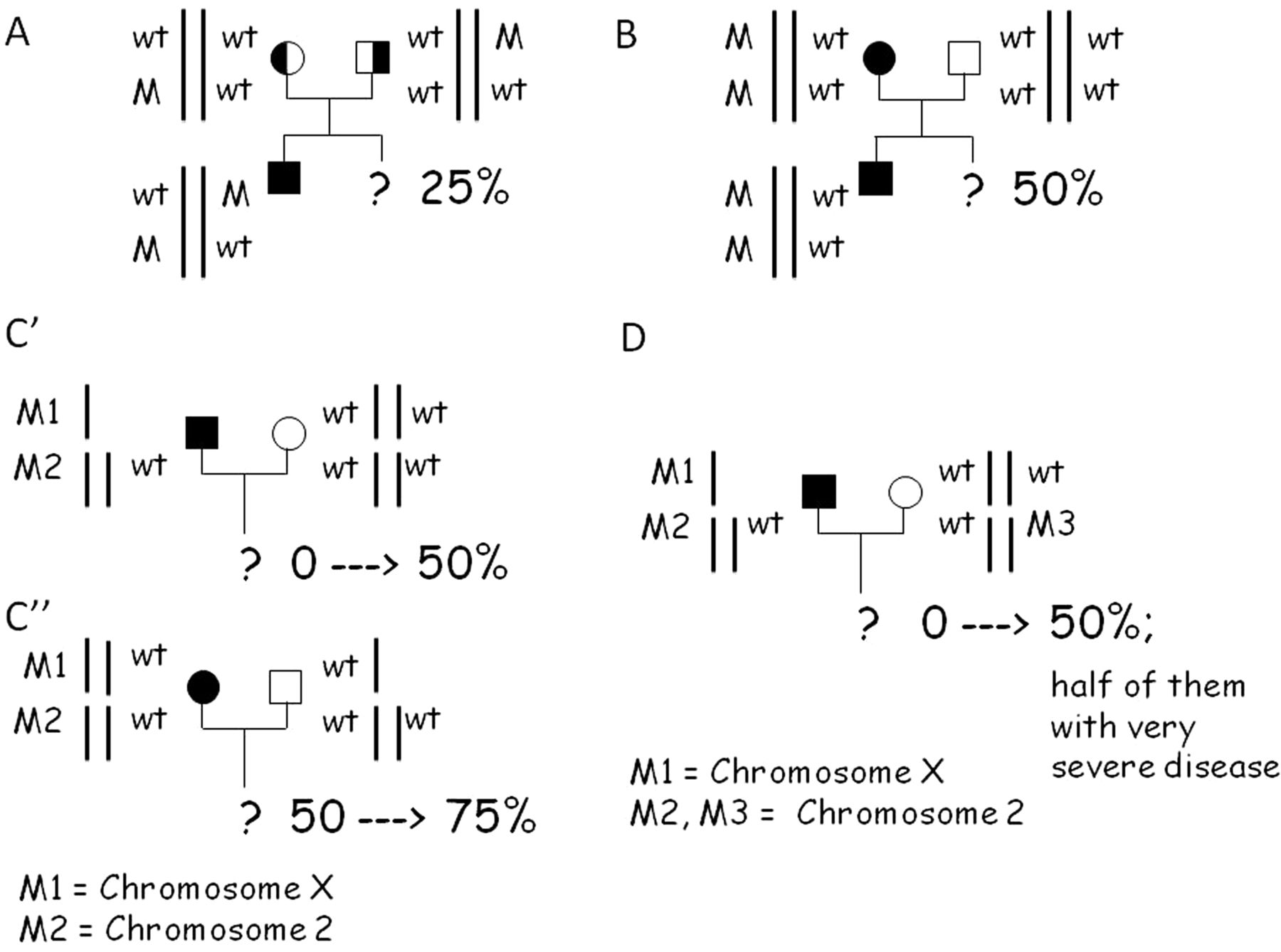
In cases of digenic inheritance in trans, the prognosis of affected individuals and the risk of recurrence for the couple overlap with those of the recessive form (figure 7A). For digenic inheritance with mutations in cis, the inheritance probability is the same as that of the autosomal-dominant disorder, but the prognosis is worse and intermediate between the autosomal-dominant and autosomal-recessive forms (figure 7B). Clinicians may need to discuss this with their patients. In digenic unlinked autosomal and X-linked inheritance, neither inheritance probability nor prognosis fits with any previously known Alport model and need to be determined on a case-by-case basis. Figure 7C′ illustrates the case of segregation through an affected man, hemizygous for an X chromosome mutation (COL4A5 gene) and heterozygous for a mutation on chromosome 2 (mutation at either the COL4A3 or COL4A4 locus). If only one mutation is detected, for example that on chromosome X, the inheritance probability of the disease is about zero. This risk may increase because of the second event reaching up to 50%. In a similar situation with mating with a heterozygote, triallelic segregation will appear in the offspring and half of them may have an even worse prognosis (figure 7D). Therefore, the present results are of interest both from a scientific point of view and for genetic counselling. Clinical geneticists should be familiar with more complex models of inheritance, which could alter the prognosis and risk of inheritance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Three possible segregation models of digenic inheritance in Alport syndrome, adapted from Schäffer.16 (A) Digenic autosomal inheritance with mutations in cis resembling a recessive inheritance disease. (B) Digenic autosomal inheritance with mutations in trans resembling a dominant inheritance. (C) Unlinked autosomal and X-linked inheritance having its own particular segregation. Panel C′ shows the variation of the risk in case of an affected father; panel C″ in case of an affected mother. (D) Trialleic inheritance as observed in family 11.
References
Footnotes
-
This work was presented at the 2014 conference of the European Society of Human Genetics where it was selected as the best scientific contribution to kidney disease by the association A.Ma.R.T.I. Onlus (Associazione Malattie Renali Toscana per l'Infanzia).
-
Acknowledgements We thank the patients and their families for permitting us to do this study. The following physicians are acknowledged for their support: Professor Grunfeld, Dr Ginglinger, Dr Dieterich and Dr Servais. Valerie Matarese provided scientific editing on parts of this manuscript.
-
Contributors All authors fulfil the criteria of authorship. AR, FF, CA, BS, KD and LH conceived and designed the study. MAM, CF and FC wrote the first draft of the paper. CF, MFA, HS, AvdW, SY and MB performed the molecular analysis. FM, HS, JAS, MvG and FF completed the final editing and revisions. All authors approved the final version. AR is the guarantor of the work.
-
Funding This work was supported in part by a donation in favour of ‘Graziano and Marco Laurini’ and Alport Syndrome Foundation (to AR) and by the Northern Counties Kidney Research Fund (to JAS). The Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases, member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, and of the EuroBioBank network, provided us with specimens.
-
Competing interests None.
-
Patient consent Informed consent for clinical data sharing and DNA testing was obtained from each proband and family member; in case of minors, parental consent was obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.