Article Text

Abstract
Background: Mutations of the filamin A locus (FLNA) on Xq28 have been established in girls with periventricular nodular heterotopia and in patients with otopalatodigital and overlapping phenotypes, the pathogenesis of these phenotypes being thought to be quite distinct. To date only six male cases of periventricular nodular heterotopia (PVNH) have been reported and these almost invariably associated with severe neurological signs.
Methods and Results: We report a new phenotype of male PVNH, with relatively normal development, no epilepsy or other neurological abnormality, severe constipation, and facial dysmorphism and without a discernible skeletal phenotype. This phenotype is associated with a splice site mutation in FLNA c.1923C>T, resulting in the generation of both normal and aberrant mRNA.
Conclusions: We postulate that the patient retains enough FLNA function to avoid the usual lethality associated with loss of function mutations in males and suggest that the severe constipation may be a clue to the molecular aetiology of other X linked conditions associated with severe constipation.
- FLNA, filamin A gene
- FMD, frontometaphyseal dysplasia
- MNS, Melnick-Needles syndrome
- OPD1, otopalatodigital syndrome type 1
- OPD2, otopalatodigital syndrome type 2
- PVNH, periventricular nodular heterotopia
- cerebro-fronto-facial syndrome
- constipation
- filamin A
- Hirschsprung disease
- periventricular nodular heterotopia
Statistics from Altmetric.com
- FLNA, filamin A gene
- FMD, frontometaphyseal dysplasia
- MNS, Melnick-Needles syndrome
- OPD1, otopalatodigital syndrome type 1
- OPD2, otopalatodigital syndrome type 2
- PVNH, periventricular nodular heterotopia
- cerebro-fronto-facial syndrome
- constipation
- filamin A
- Hirschsprung disease
- periventricular nodular heterotopia
Loss of function mutations of the human filamin A (FLNA) gene cause an X chromosomal dominantly inherited form of periventricular nodular heterotopia (PVNH), mainly observed in females.1 In heterozygous females defective initiation of neuronal migration leads to the accumulation of neurons along the walls of both lateral ventricles and is often accompanied by epileptic seizures. In affected families an excess of females as well as of miscarriages is observed, both observations pointing to intrauterine lethality of male fetuses.1,2 However, six male patients with PVNH and hemizygous FLNA mutation have been reported; these patients carry either a missense mutation or a distal truncating mutation. It was postulated that the hemizygous FLNA mutations observed in these affected males, preserve some residual protein function.3 Apart from two cases reported by Sheen et al,3 who were identified among 24 sporadically occurring males with MRI findings of PVNH, there have been four familial cases, almost all of whom have had neurological signs,4,5 most commonly epilepsy but in male patients who were essentially non-dysmorphic.
In contrast, distinct FLNA missense mutations or in frame deletions disrupt the development of craniofacial and long bones as well as additional organ systems, resulting in the allelic developmental disorders otopalatodigital syndrome types 1 and 2 (OPD1 and OPD2), frontometaphyseal dysplasia (FMD), and Melnick-Needles syndrome (MNS).6 These predominantly skeletal phenotypes of FLNA mutations are postulated to be the consequence of clustered mutations, the effect of which is to reduce the ability of filamin to sponsor actin meshwork formation.7 There is no evidence that patients with the skeletal phenotypes of FLNA mutations have a PVNH or related phenotype, although systematic evaluation for the presence of asymptomatic PVNH, to our knowledge, has not been published. Recently, Zenker et al reported a female patient presenting with a complex phenotype including clinical signs of both PVNH and FMD, resulting from a heterozygous FLNA base substitution in exon 45.8 The presence of two additional aberrant transcripts, a full length mRNA with a missense mutation (L2439M) and a shorter transcript resulting from the utilisation of a newly created splice donor site, was postulated to cause the dual phenotype in this female patient.
In this communication, we present another male patient with PVNH, associated with a splice mutation in exon 13 of the FLNA gene. In addition to PVNH, the patient also presents with craniofacial features of cerebro-fronto-facial syndrome and has severe constipation. We postulate that the predominant expression of the full length mRNA in addition to a mutant shorter transcript lacking the 3′ part of exon 13 has rescued a sufficient amount of FLNA protein function to result in this novel phenotype in a male patient hemizygous for an FLNA splice mutation. Furthermore, we suggest that our clinical observations have more widespread implications for other X linked phenotypes which merit consideration as candidate conditions for FLNA mutations.
CLINICAL REPORT
The patient is the second offspring of unrelated parents, an older brother being completely healthy. His birth weight of 3.28 kg, length of 51 cm, and OFC of 38.5 cm were not a cause for initial concern. However, he came to medical attention on day 1 of life for cardiac abnormalities, later characterised as a septal defect, pulmonary valve prolapse, and dysplastic tricuspid valve. In addition, distinct dysmorphic features were noted, including hypertelorism, down-slanting palpebral fissures, low set, posteriorly rotated ears, and talipes equinovarus (fig 1). His cardiac defects were corrected surgically, with good effect, and a later right inguinal hernia also remedied surgically. Over the ensuing 2 years, there were multiple hospital admissions for pyrexial illnesses, presumed to be viral in the absence of any positive findings to suggest otherwise. Moreover, feeding was poor, weight gain inadequate, and his parents repeatedly voiced concern about severe constipation. He was laxative dependant for bowel emptying. Investigations of his bowel function including rectal biopsy, barium follow through, and transit studies were normal. A laparotomy resulted in a Ladd procedure for a malrotation, but without meaningful effect in terms of improved bowel function. Now aged 3 years 10 months, he continues to have severe constipation, and remains laxative dependent and without apparent cause for these debilitating symptoms. His hands are normal, without evidence of spatulate fingers or proximal insertion of the thumbs.

The patient aged 2 years. The low set ears, down-slanting palpebral fissures, and hypertelorism are readily appreciated. (Photograph is published with consent.)
Meanwhile, developmental progress has been encouraging. The patient began walking at about 14 months and speaks normally for his age. His head is growing well and is consistently along the 50th centile. His height is just below the 3rd centile, as is his weight. The most recent examination confirms hypertelorism and down-slanting palpebral fissures. There have been no episodes of seizure and clinical neurological examination is normal, although it is noted that he does not frown. Normal investigations have included karyotype, skeletal survey (apart from corrected talipes), renal ultrasound, and subtelomere FISH studies. Specific negatives on examination include absence of supraorbital hyperostosis, long bone bowing, or carpal/tarsal fusion. An MRI scan at the age of 2 years showed good myelination, with heterotopic nodules of grey matter along the margins of both lateral ventricles (fig 2). The corpus callosum is normal and intact. No cortical abnormality is evident. A shallow left sided retrocerebellar cyst was noted, of doubtful significance. These findings, in the context of his facial features, led to a possible diagnosis of cerebro-fronto-facial syndrome being considered.

(A, B) Axial T2 weighted image at 3 years of age demonstrating nodular heterotopia in the myelinated brain.
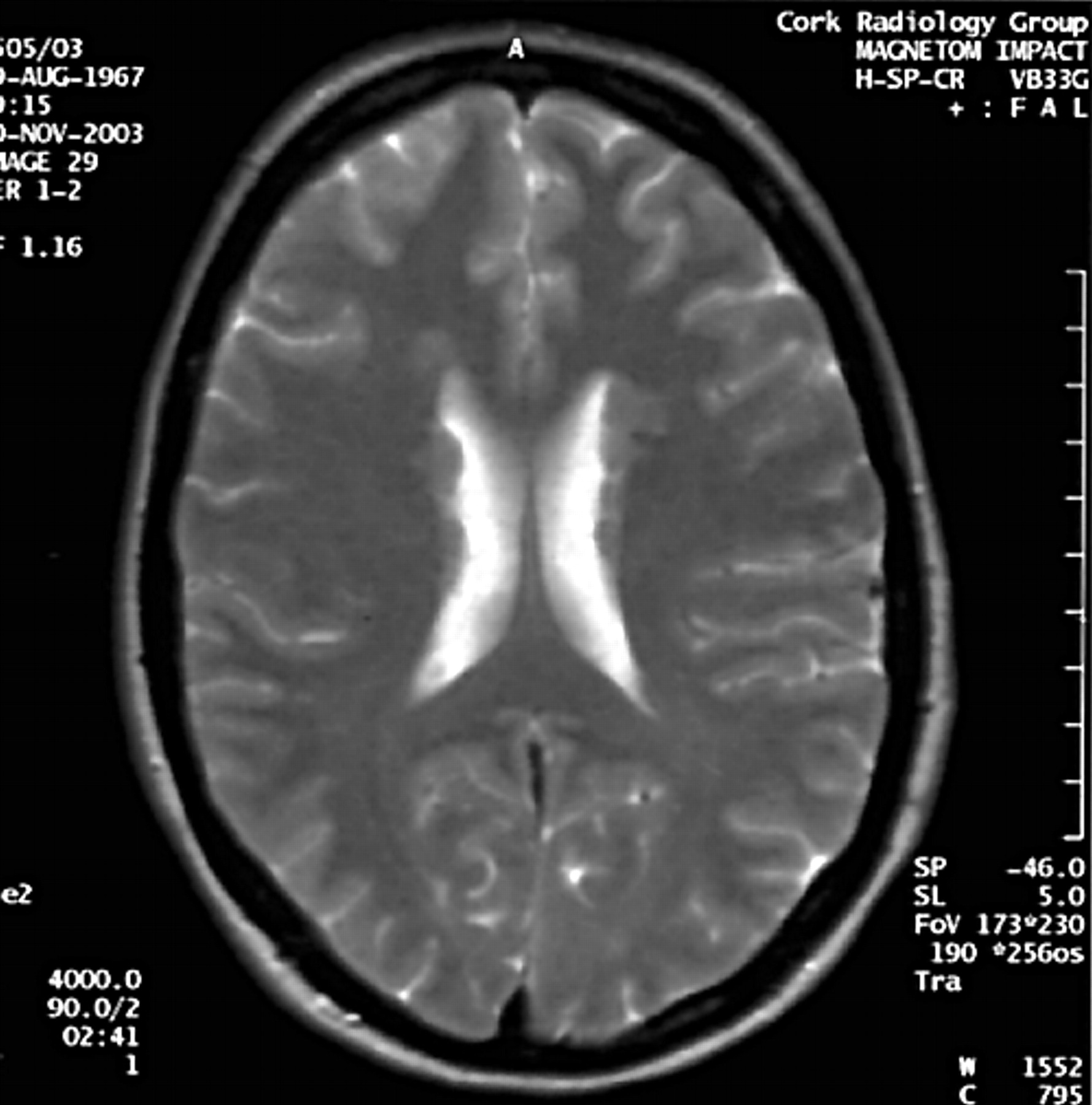
In light of these MRI findings, the family history was revisited. The mother is a clinically non-dysmorphic, intellectually normal woman of 38 years, who has never had epilepsy or other clinical evidence of CNS abnormality. MRI scan in her case shows PVNH (fig 3). Moreover, a detailed family history reveals that the maternal grandmother is epileptic, having developed symptoms in the 4th decade of life following a head injury. A maternal uncle of the proband is reported to have an unusual personality and possible psychiatric symptoms, but neither of these two individuals is available for clinical examination or MR imaging.
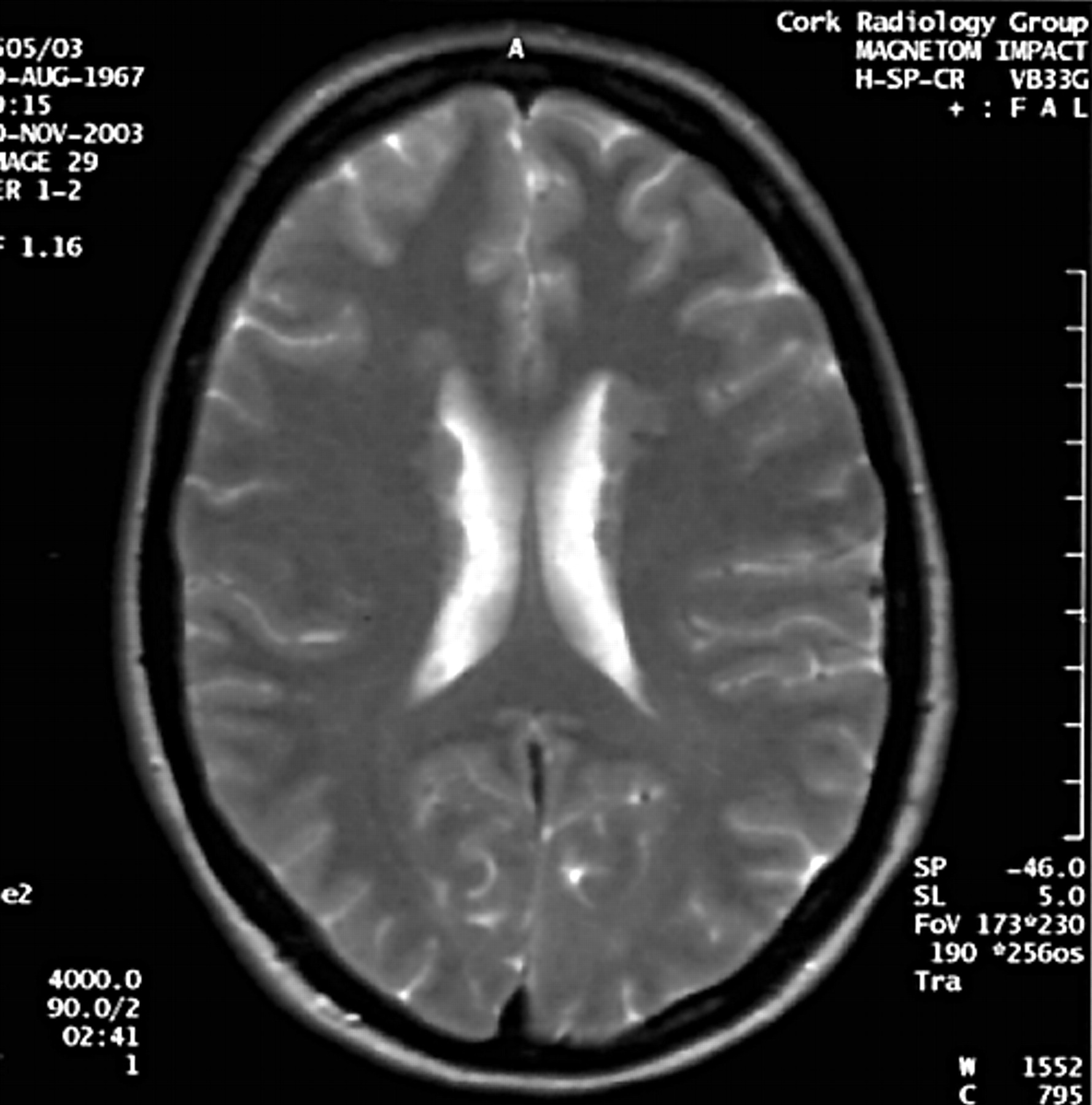
Axial T2 weighted image demonstrating nodular heterotopia along the anterior aspects of the bodies of both lateral ventricles in the mother of the proband.
METHODS
Genomic DNA was prepared from peripheral blood. The complete FLNA coding sequence including the exon-intron boundaries was amplified by PCR. Primers and reaction conditions are available on request. The amplicons were screened for mutations by direct sequencing using an ABI 3100 sequencer (Applied Biosystems, Foster City, CA). FLNA mRNA was prepared from EDTA blood by the RNAqueous kit (Ambion, Austin, TX) and transcribed into cDNA. Primers within FLNA exons 12 and 14 were used to amplify the generated cDNA.
RESULTS
Direct sequencing of the FLNA coding region revealed the index patient to be hemizygous for a base substitution in wobble position in exon 13 of the FLNA gene c.1923C>T, with both triplets coding for glycine (fig 4A,C). This base substitution is also present in his mother in heterozygous state (fig 4D), but not in the older healthy brother (fig 4B). Comparison with the consensus sequence of mammalian splice sites indicated high similarity with mammalian donor sites.9 In order to confirm a functional relevance of this base substitution, cDNA was generated from cultured lymphocytes of the index patient, his mother, and the older healthy brother. PCR amplification using primers within exons 12 and 14 resulted, for each of the three individuals, in a band of the regular size expected for the wild type allele. In addition, for both the index patient and his mother, a second smaller band was amplified. It was much weaker when compared to the intensity of the larger PCR product, but stronger for the index proband when compared to his mother (fig 4G). Sequence analysis of both bands confirmed them to be FLNA derived sequences. The smaller fragment contained the regular FLNA sequence 5′ of the base change up to the predicted position –1 of the new donor site, followed by the regular sequence of exon 14 (fig 4E,F). Alternative utilisation of this newly created donor site hence was demonstrated to result in lack of the 3′ part of exon 13 including 101 bp with frameshift and premature introduction of a stop codon in codon 681 of the aberrant mRNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Molecular genetic results demonstrating (A) the location of the base substitution in genomic DNA in exon 13 of the FLNA gene, genomic sequence of (B) the older brother, (C) the index case with hemizygous mutation, and (D) the heterozygous mother. (E) Generation of a new donor splice site. (F) Sequence analysis of the smaller PCR product demonstrates utilisation of the newly created donor splice site with lack of the 3′ part of exon 13, followed by the regular sequence of exon 14. (G) cDNA as amplified for all three family members with an additional smaller band in the index patient, which is also very faintly visible in the mother, but not in the healthy older brother.
DISCUSSION
Familial PVNH was originally described as an X chromosomal dominant trait with intrauterine lethality of affected males or early neonatal demise of those rare males who survive to birth, usually from catastrophic haemorrhage.2,10,11FLNA mutations identified to date in classical PVNH pedigrees are predicted to result in loss of function due to protein truncation.12 To date, 16 such mutations of FLNA have been reported7 widely distributed throughout the gene. Virtually all of the PVNH associated mutations are frameshift or truncating and thought to be subject to nonsense mediated mRNA decay. In contrast, all mutations described in the clinically related disorders OPD1, OPD2, FMD, and MNS, preserve the translational reading frame and result in a full length protein.7 Interestingly, in OPD2 a recognisable brain phenotype is reported including hydrocephalus and cerebellar hypoplasia, the latter also being described in a minority of PVNH patients.7
Following the description of a first female patient with heterozygous FLNA base substitution and a dual phenotype including PVNH and FMD,8 the question of further patients with intermediate phenotypes was raised.7 The report from Zenker et al8 was especially interesting, the dual phenotype in their case being occasioned by a single mutation, c.7315C>A, causing two transcripts to be produced. One such encodes a missense mutation, L2439M, while the second, the result of a cryptic splice site caused by the mutation, causes a transcript with an in-frame deletion of seven amino acids. It is postulated that the missense transcript causes the FMD phenotype, while the in-frame deletion transcript is the basis of the PVNH phenotype. With respect to intermediate phenotypes combining elements of both OPD spectrum disorders and PVNH, it is especially noteworthy that Robertson speculatively suggested cerebro-fronto-facial syndrome as a possible candidate condition for such FLNA mutation,7 although the authors have recently been advised of normal FLNA mutation analysis in respect of at least one patient reported in the literature with this condition (L Wilson, personal communication 2005).13,14 The initial working diagnosis made in relation to the subject of this report was cerebro-fronto-facial syndrome.
The facial features of the subject of this report, comprising hypertelorism, down-slanting palpebral fissures, and low set, posteriorly rotated ears are identical to those related by Robertson as typical of OPD1.7 However, the subject of this report shares none of the hand characteristics, nor the skeletal findings. Constipation is not alluded to as a feature of OPD and related spectrum disorders, but was a major clinical issue in the management of our patient. Although rectal suction biopsy showed the presence of ganglia, these were considered to be fewer in number than is usually seen, raising the possibility that neuronal migration to the enteric plexus may be incomplete in some patients with FLNA mutation. To date, reports of FLNA mutant patients are silent on this issue, but we consider that this clinical observation may be worthy of specific interrogation when obtaining their clinical history from FLNA mutation carriers. Moreover, we specifically draw attention to children with severe constipation but without histological or molecular evidence of Hirschsprung disease15 who may benefit from being considered for FLNA mutation analysis. Specifically, we observe that an X linked form of intestinal pseudo-obstruction is already described with tentative evidence of linkage to Xq28, the location of FLNA.16 Furthermore, we wonder if FLNA mutation analysis should not be offered to patients with developmental delay of varying degree and severe constipation in whom a likely clinical diagnosis of FG syndrome has been made. It may be noteworthy that a possible locus for FG syndrome (FG2) has been allocated to Xq28 on the basis of a cytogenetic inversion.17 Published expression data already support the expression of FLNA in the developing neuronal system in the wall of the digestive tract,18 consistent with the above hypothesis.
The case we present with PVNH, facially dysmorphic features, and severe constipation represents a unique clinical phenotype of FLNA mutation, overlapping in many respects with the rather ill defined condition of cerebro-fronto-facial syndrome, of which condition our case may well be an example. Of greater interest is the light which this case casts on future directions for analysis of unexplained constipation in children, for whom brain MRI and FLNA mutation analysis must surely now be considered appropriate investigations. Further delineation of FLNA associated phenotypes, such as described herein, will contribute to our understanding of the diverse functions of the normal and abnormal FLNA protein and guide the identification of further FLNA partners interacting during embryonal development.
Acknowledgments
Dr Reardon wishes to thank the family for their encouragement and kind support.
REFERENCES
Footnotes
-
Published Online First 18 November 2005
-
Dr Reardon wishes to acknowledge support from the Children’s Medical and Research Foundation for his work
-
Competing interests: none declared
-
Patient details are published with consent