Article Text

Abstract
Background: In Hirschsprung’s disease (HSCR), a hypomorphic allele of a major gene, RET, accounts for most isolated (non-syndromic) cases, along with other autosomal susceptibility loci under a multiplicative model. However, some syndromic forms of HSCR are monogenic entities, for which the disease causing gene is known.
Objective: To determine whether RET could be considered a modifier gene for the enteric phenotype on the background of a monogenic trait.
Methods: The syndromic HSCR entities studied were congenital central hypoventilation (CCHS) and Mowat-Wilson syndrome (MWS), caused by PHOX2B and ZFHX1B gene mutations, respectively. The RET locus was genotyped in 143 CCHS patients, among whom 44 had HSCR, and in 30 MWS patients, among whom 20 had HSCR. The distribution of alleles, genotypes, and haplotypes was compared within the different groups. To test the interaction in vivo, heterozygous mice were bred for a null allele of Phox2b and Ret genes.
Results:RET was shown to act as a modifier gene for the HSCR phenotype in patients with CCHS but not with MWS. The intestine of double heterozygote mice was indistinguishable from their littermates. A loss of over 50% of each gene function seemed necessary in the mouse model for an enteric phenotype to occur.
Conclusions: In CCHS patients, the weak predisposing haplotype of the RET gene can be regarded as a quantitative trait, being a risk factor for the HSCR phenotype, while in MWS, for which the HSCR penetrance is high, the role of the RET predisposing haplotype is not significant. It seems likely that there are both RET dependent and RET independent HSCR cases.
- CCHS, congenital central hypoventilation syndrome
- HSCR, Hirschsprung’s disease
- MWS, Mowat-Wilson syndrome
- SNP, single nucleotide polymorphism
- Hirschsprung’s disease
- congenital central hypoventilation syndrome
- RET
- modifier gene
Statistics from Altmetric.com
- CCHS, congenital central hypoventilation syndrome
- HSCR, Hirschsprung’s disease
- MWS, Mowat-Wilson syndrome
- SNP, single nucleotide polymorphism
Hirschsprung’s disease (HSCR, MIM 164761) is a frequent congenital malformation of the hindgut that affects 1/5000 newborn infants and is defined by the absence of enteric neurones along a variable length of the bowel.1 HSCR serves as a model in the study of diseases with a complex mode of inheritance. When isolated, the transmission is non-Mendelian with low, sex dependent penetrance. A multiplicative oligogenic model with three loci has been proposed, with RET proto-oncogene being the major gene.2 Indeed, almost all HSCR patients harbour either a heterozygous mutation of the coding sequence or, more often, a hypomorphic allele located in a conserved non-coding sequence in intron 1 and acting as a transcriptional enhancer.3,4
In about 30% of cases, HSCR is associated with other malformations. The disease causing gene is known in four syndromic HSCR forms with Mendelian inheritance: Shah-Waardenburg syndrome (MIM 277580, EDNRB, EDN3, SOX10 genes), Mowat-Wilson syndrome (MWS, MIM 235730; ZFHX1B gene), congenital central hypoventilation syndrome (CCHS, MIM209880; PHOX2B gene), and Goldberg-Shprintzen syndrome (MIM 609460; KIAA1279 gene).5–7 In each of these syndromes, penetrance for the HSCR trait is incomplete. We thus asked whether RET could be considered as a modifier gene for the enteric phenotype on the background of a monogenic trait. We tested this hypothesis in Mowat-Wilson and CCHS syndromes, where series of patients were relatively large. We show that RET acts as a modifier gene for the enteric phenotype in patients with a PHOX2B gene mutation. These data highlight the pivotal role of the RET gene not only in isolated HSCR but in syndromic HSCR as well.
METHODS
Patients
CCHS is a life threatening disorder involving an impaired ventilatory response to hypercapnia and hypoxaemia, which can be associated with other disorders of the autonomic nervous system.8 CCHS patients have HSCR in 20% of cases.9 This association is known as Haddad syndrome. MWS is a mental retardation syndrome associated with multiple congenital abnormalities which has been recently recognised as a separate entity among the heterogeneous group of patients with HSCR and mental retardation. A consistent facial gestalt allows the diagnosis of MWS in patients with no HSCR, and the penetrance of this feature is about 60%.10 When suspected, the diagnosis of HSCR is confirmed on histopathological criteria—that is, the absence of enteric plexuses and increased acetylcholinesterase histochemical staining in nerve fibres.
Molecularly proven CCHS and MWS patients were studied (fig 1). Our series of 143 CCHS patients11 included 44 with HSCR (20 female and 24 male; long segment, 22 cases; short segment, 12 cases; unknown length of the aganglionic tract, 10 cases). Our series of 30 MWS patients included 20 with HSCR (five female and 15 male; long segment, six cases; short segment, five cases; unknown length of the aganglionic tract, nine cases). We also studied a previously reported family where three members in two generations had tumours of the sympathetic nervous system of various grades at different ages (childhood in two cases and adulthood in one case).12 Most interestingly, HSCR occurred in two members of another branch of the family who were shown to carry the PHOX2B gene mutation (fig 2).

PHOX2B gene mutations in Haddad and CCHS patients and ZFHX1B gene mutations in MWS and MWS+HSCR patients. CCHS, congenital central hypoventilation syndrome; Del, deletion encompassing the gene or at least 1 exon; Fs, frameshift mutation; HSCR, Hirschsprung’s disease; Ms, missense mutation, MWS, Mowat-Wilson syndrome; Ns, nonsense mutation.
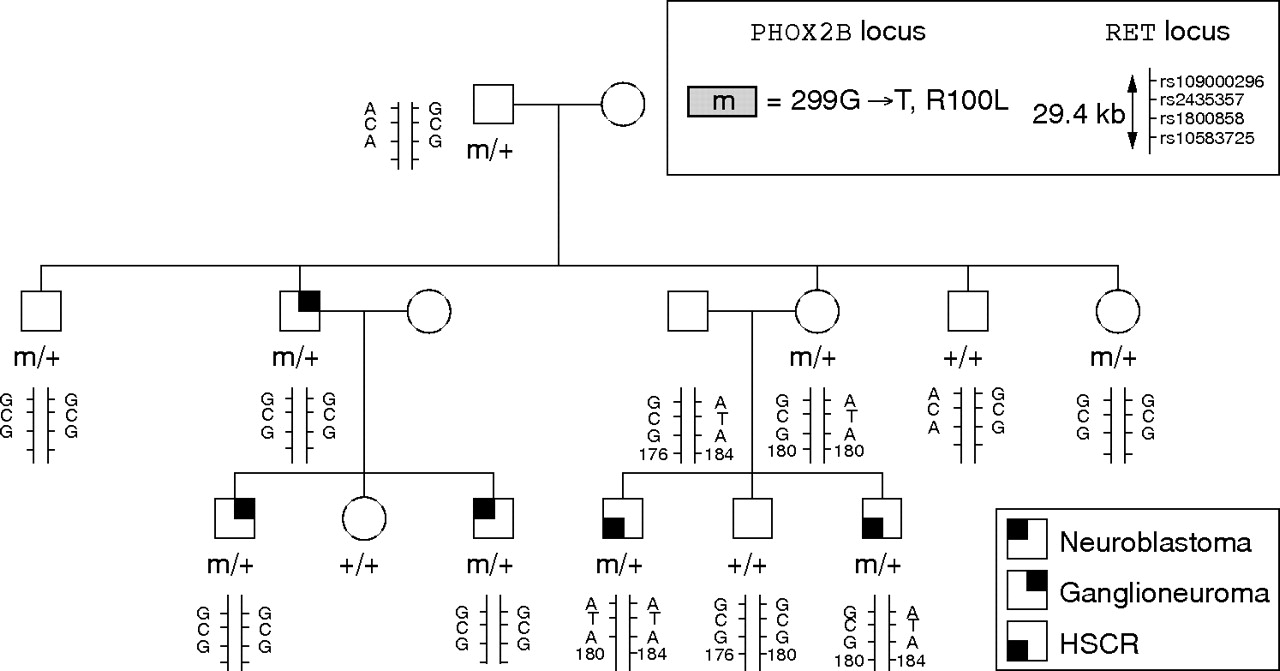
Genotypes at the PHOX2B and RET loci in family 1. Tumours of the sympathetic nervous system were found in three patients from two generations in one branch of the family, while isolated HSCR was observed in two siblings in another branch. ATA is the hypomorphic, HSCR predisposing RET haplotype and GCG the protective RET haplotype. The SNP rs10583725 is a poly AC microsatellite marker located in the intron 5 of the RET gene used to identify the parent of origin of the predisposing haplotype. HSCR, Hirschsprung’s disease; SNP, single nucleotide polymorphism.
Mouse model
We investigated whether mice heterozygous for a targeted deletion resulting in null mutations at both the Phox2b and Ret loci would present an intestinal phenotype.13,14
Methods
Blood samples were obtained with informed consent, and DNA was extracted according to standard protocols. Mutation screening of both PHOX2B and ZFHX1B genes has been described elsewhere.11,15 Patients harbouring either a heterozygous PHOX2B mutation (143 cases) or a ZFHX1B gene mutation (30 cases) were genotyped for single nucleotide polymorphisms (SNPs) rs109000296, rs2435357 and rs1800858 lying in 5′ UTR, intron 1, and exon 2 of the RET gene, respectively, in order to detect the non-syndromic HSCR predisposing haplotype (ATA).3 Direct sequencing was undertaken as previously described.16 The distribution of genotyped SNPs at the RET locus was assessed by direct counting within the different groups of patients. Allele frequencies were estimated by the counting method and genotype and allele frequencies were compared by a χ2 test. We also compared the distribution of alleles for each SNP with the nature of the PHOX2B gene mutation. Haplotypes were determined by the family studies. In addition, direct sequencing on both DNA strands of the 21 exons and flanking introns of the RET gene was carried out in the two patients with HSCR in family 1, as previously described.16
We bred Phox2b+/− females and Ret+/− males in a C57/Bl6 background.13,14 The colon of all littermates was macroscopically examined at necropsy during the second week of life. Histological examination was performed along the length of the colon from the anal margin to the caecum after haematein-eosin-safran staining.
RESULTS
The allele frequencies for each SNP tested at the RET locus were similar in both CCHS and MWS groups and remained similar to the control European population in both subgroups.3 However, the distribution of alleles varied within the CCHS group according to the presence or absence of the HSCR phenotype (table 1). In the CCHS group, the frequency of the predisposing alleles at the RET locus was 39.7% in the group of patients with Haddad syndrome (CCHS+HSCR), and 21% in the group of patients with isolated CCHS irrespective of the mutation in the PHOX2B gene (table 2). The difference was significant (χ2 = 11.73, p<0.001) and remained significant when CCHS patients harbouring an expansion of +5 alanines were excluded, as patients with CCHS and HSCR do not harbour a +5 alanine expansion (38.9% v 23.2%, p<0.02). When genotypes are considered, the odd ratios of HSCR for patients heterozygous and homozygous for the ATA haplotype were 2.39 and 4.74, respectively (table 3). In contrast, no significant differences in SNP distribution could be observed between MWS patients with and without HSCR (table 1).
Counts and frequencies (%) of the single nucleotide polymorphisms studied at the RET locus
RET haplotypes in Haddad syndrome and congenital central hypoventilation syndrome
Genotypes at the RET locus in Hadad and congenital central hypoventilation syndrome
In family 1, a heterozygous G to T transversion at nucleotide 299 in exon 2 of the PHOX2B gene and changing an arginine into a leucine in the homeodomain of the protein (R100L) segregated in patients with an autonomic nervous system disorder and the obligate carrier (fig 2). No RET gene mutation could be identified in the two patients with HSCR who inherited the PHOX2B mutation from their mother and the hypomorphic RET allele from their father (fig 2, patients III 4 and III 6). In contrast, the hypomorphic, HSCR predisposing ATA RET allele was harboured by none of the family members with no HSCR and carrying the PHOX2B gene mutation (fig 2).
Animal model
By crossing Phox2b+/− with Ret+/−, we obtained 30 pups from five mothers. All pups were killed, genotyped, and dissected. Phox2b+/−; Ret+/− mice were viable at least for the first two weeks of life (nine of the 30 pups). They showed no megacolon at necropsy. Furthermore, on histological examination, the enteric nervous system of doubly heterozygous mice was not significantly different from that of control littermates (data not shown).
DISCUSSION
Before the molecular dissection of isolated and syndromic HSCR, epidemiological data had shown obvious discordances between these conditions. Indeed, as opposed to isolated HSCR—where the sex ratio is skewed in favour of females (male to female ratio 4:1) and where the short segment form of the disease represents 80% of the cases—in Haddad syndrome, males and females are equally affected and the long segment form of the disease is the most common.1,17 This is the case in the series we report, where there are 24 males and 20 females and where long segment HSCR occurred in the majority of cases for which the length of the aganglionic tract was known. However, it is worth mentioning that among patients presenting with an isolated, long segment form of HSCR, skewing of the sex ratio decreases when compared with the short segment form of HSCR. In relation to MWS, we observed a skewed male to female ratio of 2.5:1 among the 20 patients with HSCR (15 males, five females), in agreement with previous reports.10
RET had been considered as a candidate gene for CCHS and heterozygous missense mutations were identified in four Haddad cases and one CCHS case, respectively.18–22 Such mutations were found in a minority of patients and were inherited from an asymptomatic parent each time it could be tested. Subsequently, PHOX2B has been identified as the major disease causing gene in CCHS with a heterozygous de novo mutation in over 90% of the cases.11,23,24 Frameshift and missense mutations lying in the homeodomain of the protein are rare (11% in this series, fig 1) and predispose to tumours of the sympathetic nervous system.11 The most frequent PHOX2B gene mutation is an in-frame duplication resulting in a de novo expansion of +5 to +13 alanines in a row of 20 alanines C-terminal to the homeodomain of the protein (fig 1).11 The distribution of alanine expansions differs among subgroups of patients, as the smallest alanine expansion (+5 alanines) is never found in Haddad syndrome cases (fig 1).11,23,24 However, when considering +6 and +7 alanine expansions, 19% and 33% of the patients, respectively, have HSCR in addition to CCHS. Therefore, for an identical PHOX2B gene mutation, patients present CCHS with or without HSCR. Such a finding clearly indicates a role for modifier factors in the occurrence of the HSCR phenotype—allelic diversity is not the only satisfactory explanation. Concerning MWS, ZFHX1B mutations are either de novo heterozygous deletions encompassing the gene or truncating mutations, both leading to a loss of function.25
A PHOX2B gene mutation appears necessary and sufficient for the CCHS phenotype to occur. Here we show that the RET gene acts as a modifier of the PHOX2B gene. Indeed, the frequency of the ATA haplotype of the RET gene is similar to that observed in the European population (25%)3 in all groups except the Haddad group, where its frequency reaches 38.6% (χ2 = 11.73, p<0.001). However, a strict digenic model does not account for all cases and at least one more modifier gene (with or without the involvement of environmental factors) must be involved. Indeed, 16 patients with a PHOX2B alanine expansion and no predisposing RET haplotype had HSCR. Among these cases, one is known to harbour a heterozygous RET gene missense mutation P1039L,19 in addition to a de novo +7 alanine expansion of PHOX2B. One can hypothesise that further candidate modifier genes in CCHS could be either genes involved in the PHOX2/RET developmental cascade or genes yet to be found at the 3p21 and 19q12 loci in isolated HSCR.
In mice, the paired-like homeobox 2b (Phox2b) gene is a key player in the development of neurones of the autonomic nervous circuit.13,26,27 Homozygous knock-out mice die in utero on day E13.5; most neurones of the autonomic reflex circuit either fail to form or degenerate and Phox2b is required for Ret to be expressed in migrating enteric neuroblasts. Phox2b−/− and Ret−/− mice have no enteric nervous system, while no megacolon has been observed in either Phox2b+/− or Ret+/− mice.13,14,28 Therefore, a haploinsufficiency of 50% at the Phox2b or the Ret locus is not sufficient for an abnormal development of the enteric nervous system in mice as opposed to what is observed in humans.1,29 We observed no abnormality of the intestine of Phox2b+/−; Ret+/− mice either macroscopically or microscopically. We may have to await the generation of mouse models where Phox2b, Ret, or both proteins have a less than 50% residual activity. No hypomorphic Ret allele is known in mice, and a knock-in mouse model leading to a +7 alanine expansion of Phox2b is being generated.
The role of polymorphisms as modifiers of a disease phenotype is being increasingly recognised. Interestingly, Phox2b has recently been considered a putative modifier gene in Dom mice when considering the megacolon phenotype.30Sox 10, a member of the Sry family, is mutated in Dom, and its human homologue is disease causing in autosomal dominant Shah-Waardenburg syndrome. A similar effect will be difficult to test in humans as a SOX10 mutation is rare while the penetrance of HSCR is high in carriers. However, the putative role of the RET gene could be further tested in syndromic HSCR entities frequent enough to allow the collection of large series of patients, namely cartilage-hair hypoplasia, Bardet-Biedl syndrome, and Down’s syndrome. In relation to the MWS group of patients, we did not observe a significant difference in allelic distribution at the RET locus between patients with or without HSCR. Nevertheless, the number of patients tested thus far remains small, and we cannot exclude the possibility that RET could still play a modest role in the HSCR phenotype of MWS patients. If not, the ZFHX1B gene would then be the first entirely “RET independent” gene responsible for the HSCR phenotype. Interestingly, Zfhx1b is an intranuclear regulator of the transforming growth factor β signalling pathway, expressed in premigratory cranial and vagal neural crest cells in mice.31
Our data argue that in man RET is a modifier gene in the context of a PHOX2B gene mutation for the HSCR phenotype, with a relative risk ranging from 2.4 to 4.7 in heterozygotes and homozygotes for the RET predisposing haplotype, respectively. These data show interactions between genes involved in the same developmental cascade and extend the concept of multistep control well known in metabolic pathways to a developmental pathway (fig 3). To our knowledge, similar examples remain uncommon. One worth mentioning is Alagille syndrome caused by heterozygous mutations of JAG1, a ligand for the Notch family of receptors. Jag+/− mice have a subtle ocular phenotype, while mice doubly heterozygous for a null allele of Jag1 and a hypomorphic allele of Notch2 have a more severe complex phenotype closely resembling Alagille syndrome.32 In HSCR, genetic interactions between RET and genes encoding proteins of the endothelin signalling pathway (EDNRB and ET3) have been documented in both humans and mice.33–35 Altogether, RET plays a pivotal role in HSCR as the major gene in isolated HSCR and as a modifier gene in at least some syndromic HSCR entities.
{kind=link}
{kind=link}
{kind=link}
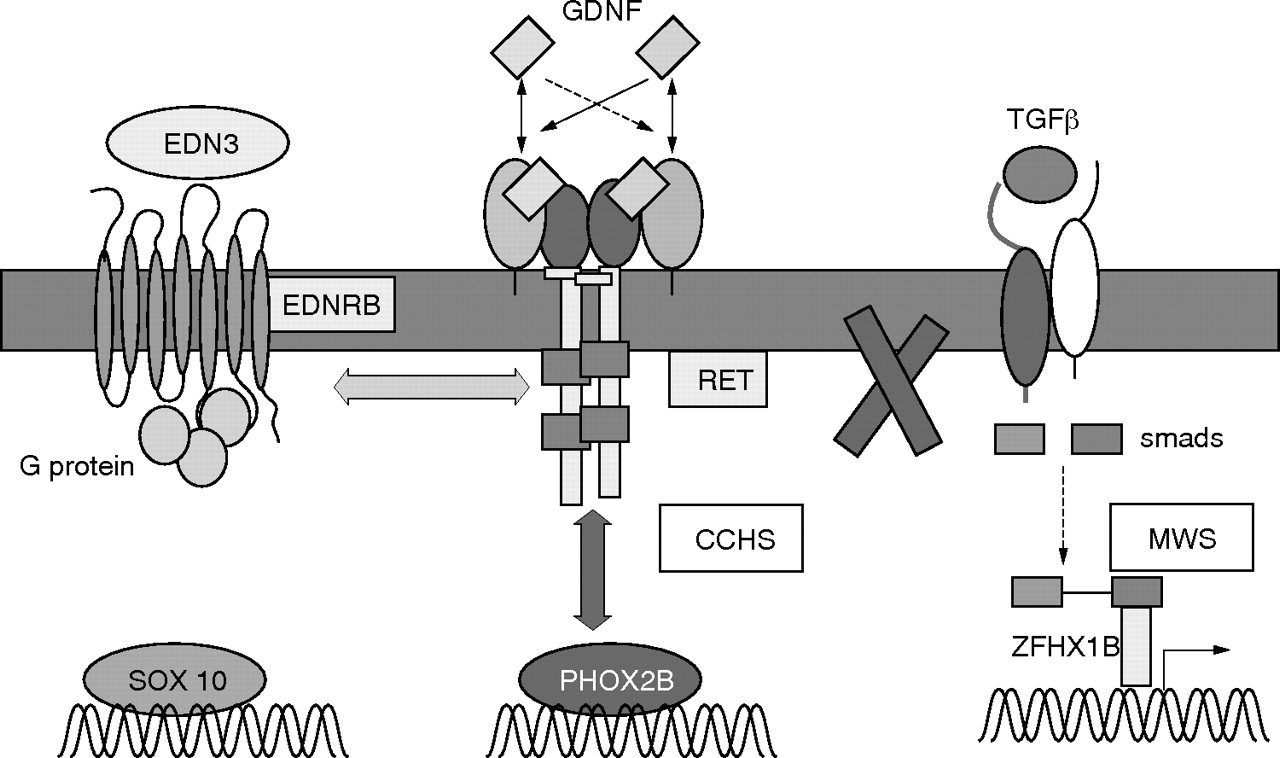
Signalling pathways involved in isolated and syndromic Hirschsprung’s disease. The figure shows the three main developmental pathways involved in the normal enteric nervous system development and in isolated and syndromic Hirschsprung’s disease. The RET pathway interacts in humans and mice with the endothelin pathway but not with transforming growth factor β (TGFβ) pathway. EDNRB (endothelin receptor type B) acts as a modifier in Dom SOX10 mutant mice. This study shows that RET interacts with PHOX2B in CCHS patients. Conversely, no genetic interaction could be found in MWS. CCHS, congenital central hypoventilation syndrome; MWS, Mowat-Wilson syndrome.
Acknowledgments
We thank the families and their referring clinical teams for participation and the Association Française de la Maladie de Hirschsprung for its support. This work was supported by grants from the GIS-Maladies Rares and the Association Nationale pour la Recherche.
REFERENCES
Footnotes
-
Published Online First 27 January 2006
-
Conflicts of interest: none declared