Article Text

Abstract
Background: Human autosomal recessive primary microcephaly (MCPH) is a heterogeneous disorder with at least six genetic loci (MCPH1–6), with MCPH5, caused by ASPM mutation, being the most common. Despite the high prevalence of epilepsy in microcephaly patients, microcephaly with frequent seizures has been excluded from the ascertainment of MCPH. Here, we report a pedigree with multiple affected individuals with microcephaly and seizures.
Objective: To identify the gene responsible for microcephaly and seizures in this pedigree.
Methods: Clinical assessments of three patients and brain MRIs of two patients were obtained. Genome-wide linkage screen with 10 k SNP microarray, fine mapping with microsatellite markers, and mutational analysis of the genomic DNA were performed on the pedigree.
Results: We found that the family was linked to the MCPH5 locus on chromosome 1q31.2–q32.1. We screened ASPM and identified a previously unreported nonsense mutation that introduced a premature stop codon in exon 18 of the ASPM gene.
Conclusions: We thus expand the clinical spectrum of ASPM mutations by showing that they can occur in patients with seizures and that the history of seizures alone should not necessarily preclude the diagnosis of primary microcephaly.
- HC, head circumference
- MCPH, autosomal recessive primary microcephaly
- SD, standard deviation
- ASPM
- epilepsy
- mental retardation
- microcephaly
- seizure
Statistics from Altmetric.com
A major cause of mental retardation is microcephaly (“small head”), in which the head circumference (HC) is reduced because of a congenital deficiency of fetal brain development, particularly affecting the cerebral cortex.1,2 Microcephaly has a heterogeneous etiology with environmental and genetic factors.3–5 The prevalence of epilepsy is high in microcephaly patients.6,7 Familial microcephaly is predominantly autosomal recessive.6 Autosomal recessive primary microcephaly (MCPH; MIM 251200) is clinically defined as congenital microcephaly with an HC at least three standard deviations (SDs) below the expected mean for age and sex, with mental retardation but no other neurological, growth, health, or dysmorphic findings, and no discernible prenatal or postnatal syndrome or cause, such as chromosome anomalies or structural brain anomalies.8 In MCPH patients, seizures are considered to be no more frequent than in the general population.9 Genetic heterogeneity of MCPH has been shown, with six loci (MCPH1–6) reported to date, all of which are clinically indistinguishable disorders.10–17 Mutations in MCPH1 (microcephalin; MIM 607117) on chromosome 8p23 and in ASPM (abnormal spindle-like, microcephaly-associated; MIM 605481) on chromosome 1q31.3 have been identified to cause MCPH1 and MCPH5, respectively, with ASPM mutations being the most common cause of MCPH.9,18–21 Despite the high prevalence of epilepsy in microcephaly patients, patients with frequent seizures have been excluded from the ascertainment of MCPH. As a result, no mutations in known MCPH genes (MCPH1 and ASPM) have been reported in patients with microcephaly and epilepsy to date. Here we report the first ASPM mutation identified in patients with microcephaly and seizures in a consanguineous family, suggesting that either ASPM mutations can cause disorders beyond MCPH, or that the spectrum of MCPH should be broadened to include seizures.
METHODS
The consanguineous family originated from Saudi Arabia (fig 1). The first cousin parents had six unaffected children and three children with microcephaly, who were 18.5, 15, and 3.5 years of age, respectively, at the time of latest examination. The HCs of the three affected children were 4 to 5 SDs below the age and sex matched population mean.22 All three affected children had normal motor development. However, IV:1 had severe mental retardation with an IQ of 35, whereas IV:3 showed moderated mental retardation with an IQ of 55.

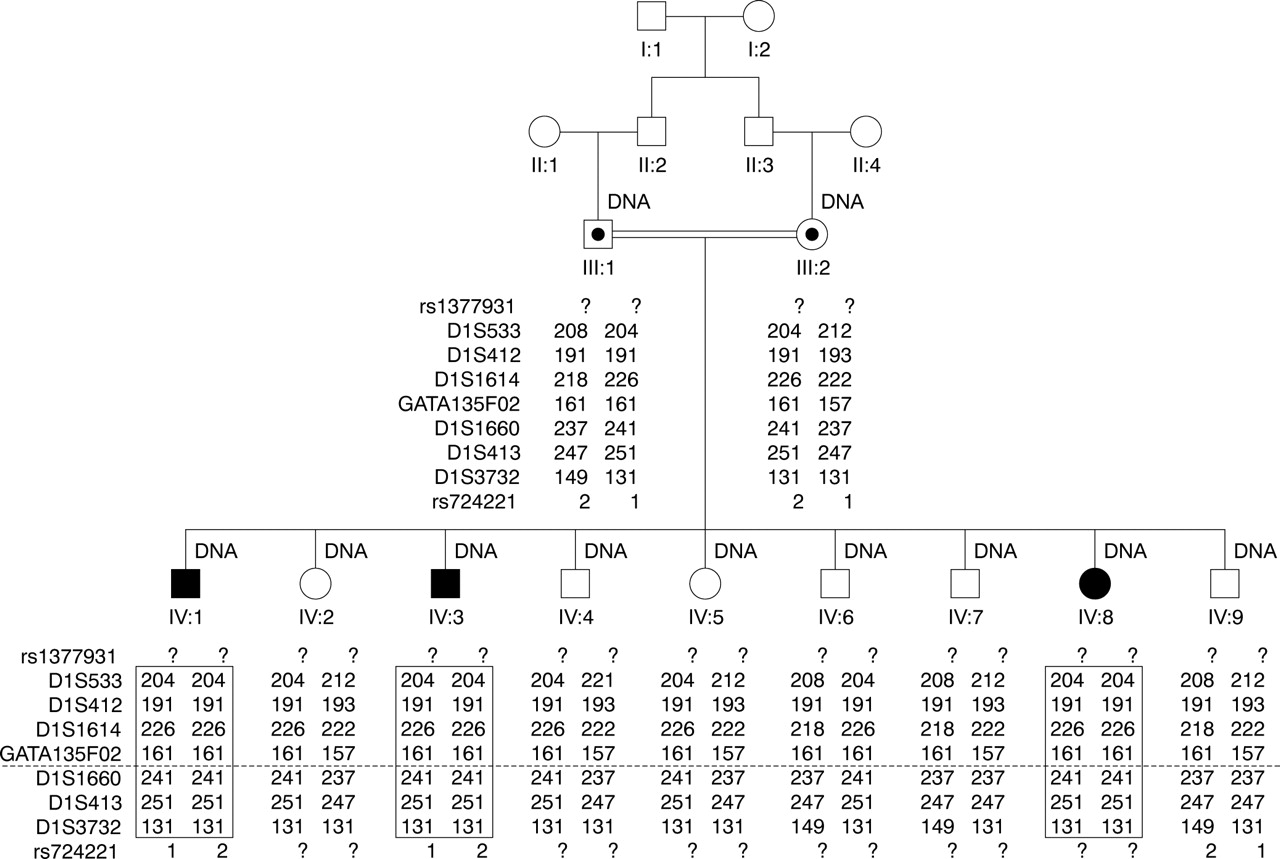
The pedigree of the family with microcephaly and seizures. IV:1, IV:3, and IV:8 had microcephaly. IV:1 and IV:8 also had seizures. DNA samples were available for individuals labelled “DNA”. The genotypes of seven microsatellite markers between the two SNP markers near the MCPH5 locus at chromosome 1q31.2–q32.1 are shown. The marker order is cen-rs1377931-D1S533-D1S412-D1S1614-GATA135F02-[MCPH5]-D1S1660-D1S413- D1S3732-rs724221-tel. “?” indicates that the allele information is not available. The dotted line indicates the location of the MCPH5 gene, ASPM. The boxed regions indicate the homozygous haplotype shared among all three affected children.
IV:1 and IV:8 had frequent tonic/clonic seizures with onset at 30 and 32 months of age, respectively. Their seizures had no other obvious causes, such as fever or head injury. The EEG of IV:1 at 3 years of age showed bilateral dysrhythmia and grade 3 rolandic spikes. His seizures started as transient paroxysmal attacks with right sided hemiparesis lasting for 5 min, after which he would fall down or sometimes lose consciousness. At 11 years old, his EEG showed left sided epileptic discharges. His seizures occurred 1–2 times per week until he was 16 years old, when they were controlled by medication (carbamazepine). He has been free of seizures over the last 2 years. IV:8 was noted to wake up from sleep scared with left sided stiffness lasting about 1 min. These attacks took place 3–7 times per night. Her EEG at 3 years of age showed slowing of background activity at 5 Hz, with an amplitude of 20–100 μV, during the awake state and abnormal right central spikes especially during the sleep portion of the recording. The seizures of IV:8 were not under control by medication.
The brain MRIs of IV:3 and IV:8 showed no structural malformation except for small brain size (fig 2). The cerebral cortical gyral pattern was mildly simplified and the volume of the white matter was slightly reduced, consistent with the small size of the brain. The fibre tracts showed normal development and myelination. There were no heterotopia or other abnormalities. The reduction in brain size appeared to be fairly uniform without preferential involvement of particular brain regions. The clinical information was consistent with MCPH with the exception of the history of severe seizures in two of the three affected children.

Brain MRIs of two affected individuals (IV:3 and IV:8) with and without seizures. (A) Axial T1-weighted image from IV:3, who had no seizure, taken at 10 years of age. (B) Sagittal T2-weighted image from IV:3 taken at 10 years of age. (C) Axial T1-weighted image from IV:8, who had recurrent intractable seizures, taken at 12 months of age. (D) Sagittal T1-weighted image from IV:8 taken at 12 months of age.
DNA was extracted from peripheral blood lymphocytes by means of a standard non-organic extraction procedure (Qiagen, Valencia, CA). A genome-wide screen for regions of linkage was performed with Affymetrix 10 k SNP GeneChip analysis on all three affected children (IV:1, IV:3, and IV:8), one unaffected sibling (IV:9), and the parents (III:1 and III:2) according to the manufacturer’s specifications (Affymetrix, Santa Clara, CA). For fine mapping, we selected seven polymorphic microsatellite markers surrounding the MCPH5 locus at chromosome 1q31.3–q32.1 from UCSC Human Genome Browser and had the primers synthesised (Invitrogen, Carlsbad, CA). PCR reactions were performed in 7.5 µl volumes that contained 10 ng genomic DNA; 0.33 µM primers; 250 µM each dGTP, dCTP, dTTP, and dATP (Invitrogen, Carlsbad, CA); 1×reaction buffer; and 5 U HotStar Taq DNA polymerase (Qiagen). Amplified markers were electrophoresed on the ABI Prism 3100 Genetic Analyzer using standard fragment analysis protocols according to the manufacturer’s specifications (Applied Biosystems, Foster City, CA). We performed fragment-length analysis using the ABI Prism Genescan and Genotyper 3.7 analysis packages. Two-point and multi-point linkage analysis was performed using Allegro version 1.2.23 A fully penetrant autosomal recessive mode of inheritance and equal allele frequencies of the markers were assumed. The disease allele frequency was estimated at 1/1000. We detected all mutations by bi-directional sequence analysis of genomic DNA using standard methods. The exons and flanking intron regions were amplified by PCR using primers previously described.20 The amplified PCR products were sequenced by automated fluorescent dye terminator methods on ABI Prism 3730xl sequencers (SeqWright, Houston, TX).
RESULTS
A single 10.37 Mb region of shared homozygosity and allele sharing among the three affected individuals was identified at chromosome 1q31.2–q32.1 between markers rs1377931 and rs724221 by Affymetrix 10 k SNP GeneChip analysis, overlapping the MCPH5 locus. No other regions of suggestive linkage were seen in the genome. Further refinement of the region by microsatellite marker analysis confirmed linkage to MCPH5. All affected individuals were homozygous for the same haplotype in the region (fig 1). The maximum two point LOD score was 2.43 at all informative markers within the region with θ = zero, and the maximum multipoint LOD score was 3.03 at the locus. Mapping of the disease locus in this family to MCPH5 implied that ASPM might be the causative gene, although the finding that two of the three affected children had seizures might suggest the disease is distinct from MCPH5.
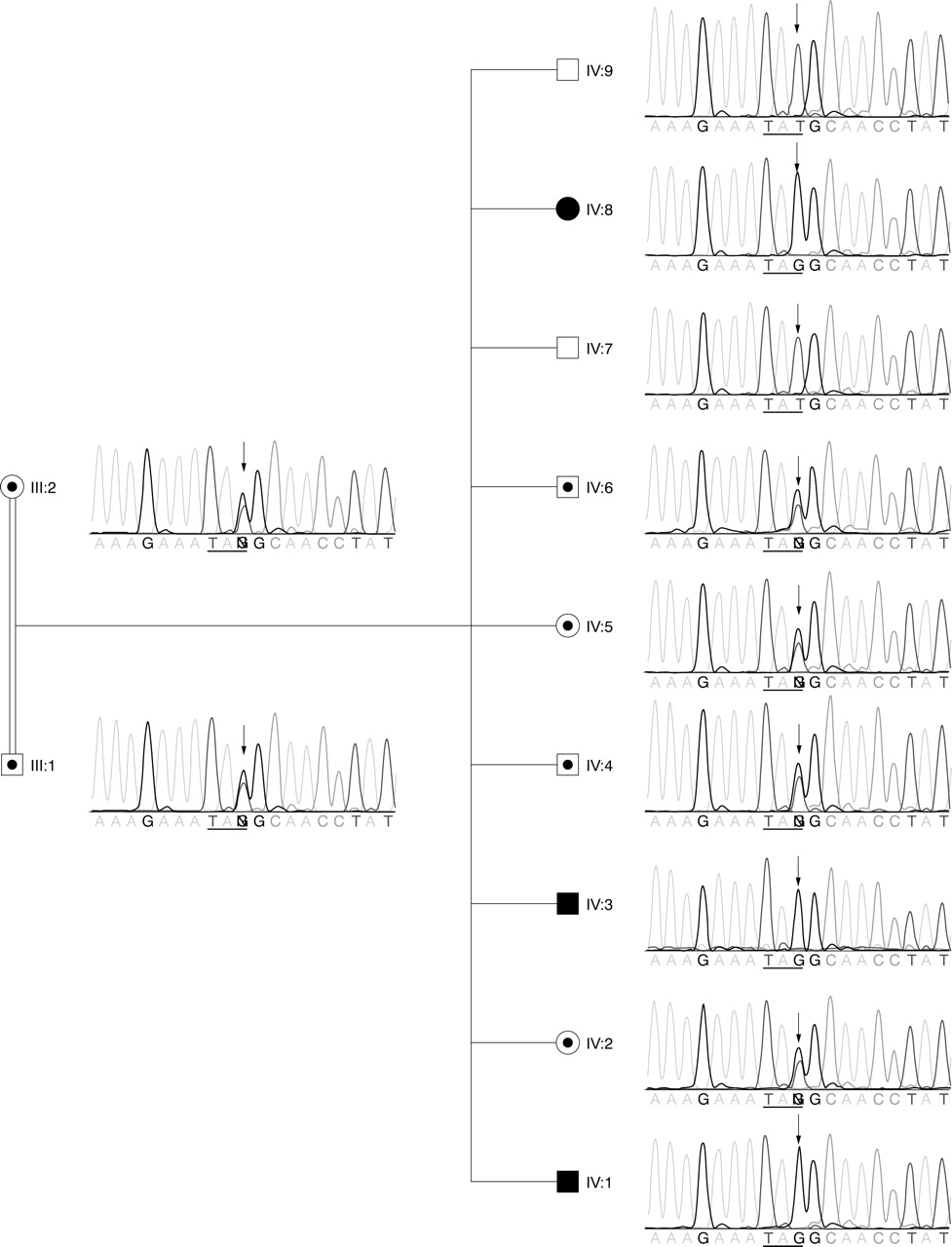
To determine whether ASPM might be the causative gene, we performed mutation analysis by sequencing the 28 exons and flanking intron regions of the ASPM gene in all members of the family. A previously unreported homozygous 6189T→G transversion in exon 18 was identified in all three affected individuals (fig 3). This mutation changes the tyrosine-2063 (TAT) to a premature amber stop codon (TAG). The mutation segregated with the haplotype. In addition, we also identified four non-synonymous (7480T→C, 7684A→G, 7939C→A, and 9395T→G) and six synonymous (849C→T, 3579T→A, 4449A→G, 5961A→G, 7566A→G, and 7674C→T) SNPs in the exons. Six of the SNPs (rs964201, rs3762271, rs6677082, rs4915337, rs2878749, and rs1412640) were described in the dbSNP database, and the other four SNPs were previously reported to occur in individuals with MCPH.9
{kind=link}
{kind=link}
{kind=link}
The 6189T→G (Y2063X) mutation in ASPM identified in patients with MCPH and seizures. The chromatographs of the DNA sequencing results are shown next to the symbols of the individuals. The arrows point to the nucleotide position 6189T in ASPM, which was mutated to “G” in the three affected children. The parents were heterozygous. IV:2, IV:4, IV:5, and IV:6 were heterozygous carriers. IV:7 and IV:9 were homozygous for the normal allele. The horizontal lines underline the codon. The normal tyrosine codon (TAT) was changed to an amber stop codon (TAG) in the mutant allele.
DISCUSSION
We report here the identification of the first ASPM nonsense mutations in patients with microcephaly and seizures. ASPM and MCPH1 mutations have been reported to associate with MCPH, but none of the patients with ASPM or MCPH1 mutations have been reported to have seizures to date. MCPH has been historically ascertained without other neurological abnormalities including frequent seizures, so that microcephaly with seizures has been generally considered to have a genetic cause distinct from that of MCPH. The brain MRIs of the patients described here were consistent with MCPH. However, it was unusual to observe that two of the three MCPH patients had frequent seizures. Our results provide the first evidence that epilepsy can occur in patients with ASPM mutations.
A total of 24 mutations in the ASPM gene have been reported in MCPH patients.9,20,21 These mutations are all protein truncating mutations and are scattered throughout the gene except for a ∼3 kb gap (nucleotide position 4796–7760) in the 4755 bp exon 18. Interestingly, the 6189T→G mutation we identified in patients with seizures is also a protein truncating mutation, but it falls within the previous mutation free zone. The 6189T→G mutation may induce nonsense mediated decay of the mRNA or result in a 40% truncation of the 3477 amino acid full length protein. Since one of our patients with the same mutation did not have seizures, there appeared to be no correlation between the mutation and the phenotype. On the other hand, it is unlikely that the seizures were due to a second genetic locus in the family, because there was no family history of seizures. Therefore, the manifestations of seizures in two of the three microcephaly patients may suggest that patients with the 6189T→G (Y2063X) mutation in ASPM are more susceptible to seizures, perhaps due to an allelic specific effect.
Alternatively, the occurrence of seizures in MCPH patients may generally have been underestimated, because of the strict “no seizures” criterion of the definition of MCPH. Therefore, it is possible that other microcephaly patients with seizures may also have undetected mutations in ASPM or MCPH1. Therefore, perhaps the phenotypic spectrum of MCPH should be broadened to include seizures, and seizure patients who otherwise fit the definition of MCPH should be advised to have mutation testing in known MCPH genes. Given the high prevalence of seizures in microcephaly patients, a large number of families with microcephaly and seizures might have been systematically excluded from linkage studies of MCPH. Including these families will help to refine the mapping of other MCPH loci, where the responsible genes have not been identified.
ACKNOWLEDGEMENTS
We are grateful to the family who kindly consented to join the study. We thank the Microarray Core Facility at Dana Farber Cancer Institute (Boston, MA) for help with GeneChip hybridisation and data acquisition. We thank Kathryn Allen for excellent technical support, R Sean Hill for help with using the linkage analysis software, and other members of Walsh Laboratory for kind help and advice.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work.
-
This work was supported by National Institutes of Neurological Disorders and Stroke grant R37NS035129 (to CAW). JS is a Stuart HQ and Victoria Quan Fellow. CAW is an Investigator of Howard Hughes Medical Institute.
-
Competing interests: none declared. The funding sources had no direct involvement in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
-
Ethics approval: The genetic study was approved by Beth Israel Deaconess Medical Center Institutional Review Board. Appropriate informed consents were obtained from all involved human subjects.