Article Text

Abstract
Background: A new syndrome has been recognised following thorough analysis of patients with a terminal submicroscopic subtelomeric deletion of chromosome 9q. These have in common severe mental retardation, hypotonia, brachycephaly, flat face with hypertelorism, synophrys, anteverted nares, thickened lower lip, carp mouth with macroglossia, and conotruncal heart defects. The minimum critical region responsible for this 9q subtelomeric deletion syndrome (9q−) is approximately 1.2 Mb and encompasses at least 14 genes.
Objective: To characterise the breakpoints of a de novo balanced translocation t(X;9)(p11.23;q34.3) in a mentally retarded female patient with clinical features similar to the 9q− syndrome.
Results: Sequence analysis of the break points showed that the translocation was fully balanced and only one gene on chromosome 9 was disrupted—Euchromatin Histone Methyl Transferase1 (Eu-HMTase1)—encoding a histone H3 lysine 9 methyltransferase (H3-K9 HMTase). This indicates that haploinsufficiency of Eu-HMTase1 is responsible for the 9q submicroscopic subtelomeric deletion syndrome. This observation was further supported by the spatio-temporal expression of the gene. Using tissue in situ hybridisation studies in mouse embryos and adult brain, Eu-HMTase1 was shown to be expressed in the developing nervous system and in specific peripheral tissues. While expression is selectively downregulated in adult brain, substantial expression is retained in the olfactory bulb, anterior/ventral lateral ventricular wall, and hippocampus and weakly in the piriform cortex.
Conclusions: The expression pattern of this gene suggests a role in the CNS development and function, which is in line with the severe mental retardation and behaviour problems in patients who lack one copy of the gene.
- ISH, in situ hybridisation
- MLPA, multiplex ligation dependent probe amplification
- 9q
- Eu-HMTase1
- mental retardation
- subtelomeric deletion
Statistics from Altmetric.com
Submicroscopic telomeric deletions account for about 5% of the patients with unexplained mental retardation.1–3 This figure is dependent on the patient selection used for studying such aberrations, and ranges from 0.5% in mild mental retardation to 7.4% in children with moderate to severe retardation.4 Some submicroscopic telomeric deletions are associated with a specific phenotype, such as 1p−, 4p−, 5p−, 9p−, 18p−, and 17p−.2,3 For most subtelomeric deletions a specific phenotype has not yet been defined, mainly because of the paucity of reported cases. The 9q34.3 deletion is emerging as one of the more common telomeric deletions and the phenotype is gradually becoming better defined.
Up to now, 22 patients have been reported with a submicroscopic subtelomeric deletion of 9q.2,4,5,6,7,8,9,10,11 The phenotypic features commonly seen in these patients are severe mental retardation, hypotonia, microcephaly, flat face with hypertelorism, synophrys, short nose with anteverted nares, thickened lower lip and carp mouth with tongue protrusion, and conotruncal heart defects. Less frequently observed features are obesity, hearing loss, urogenital abnormalities, and behavioural problems. Stewart et al8 defined the critical chromosomal region responsible for this phenotype by mapping the deletion end points in 12 patients with a subtelomeric deletion of chromosome 9qter. This minimum critical region is approximately 1.2 Mb, encompassing at least 14 genes or transcripts, and it is suggested that haploinsufficiency of one or more genes within this region most probably accounts for the 9q− syndrome.
We have previously reported the characterisation of the breakpoints of a de novo balanced translocation t(X;9)(p11.23;q34.3) present in a female patient with severe mental retardation.12 The translocation was found to be completely balanced at the DNA level and disrupted the ZNF81 gene on the X chromosome, while on chromosome 9 the break point disrupted the Eu-HMTase1 gene. The ZNF81 gene was subsequently screened for mutations in a cohort of over 300 male patients with X linked mental retardation. A missense mutation in the ZNF81 gene was identified in a linked family with mild non-specific mental retardation.12 However, the non-syndromic phenotype in this family is clearly different from that of the translocation patient, indicating that the disruption of ZNF81 is not causative for the severe clinical features in this patient.
In this paper we report a careful evaluation of the clinical presentation of the t(X;9) patient, which revealed a striking phenotypic overlap with the 9q− syndrome. The chromosome 9 breakpoint in this patient disrupts the Eu-HMTase1 gene, which precisely maps to the common deletion region found in the 9q− syndrome. The specific disruption of Eu-HMTase1 suggest that haploinsufficiency of this gene is responsible for the larger part of the 9q− phenotype.
METHODS
Subjects
Index patient
The index patient had been referred to our department for genetic diagnosis. Routine cytogenetic studies revealed a karyotype that showed a translocation t(X;9)(p11.2;q34.3). This translocation appeared to be de novo because both parents had a normal karyotype.
Patient panel
To search for aberrations we selected 37 mentally retarded patients with a normal karyotype but with facial features seen in the 9q− syndrome and previously screened routinely for submicroscopic telomeric deletions within our department. Genomic DNA of each patient was isolated using standard procedures.13
Multiplex ligation dependent probe amplification
A specifically designed set of probes for testing for subtelomeric chromosomal imbalances—SALSA P036 human telomere test kit (MRC-Holland, Amsterdam, Netherlands)—was used for subtelomere screening14 and included one probe in the MRPL41 gene at 9q34. In addition, a specifically designed probe set including synthetic probes for the 9q subtelomeric region was developed. Probe preparation has been described previously (see electronic database information). The multiplex ligation dependent probe amplification (MLPA) mix contained two probes for CACNA1B and two for Eu-HMTase1. The full sequences of these probes are available on request.
MLPA analysis was carried out for all 37 selected patients using standard kits, as described by Schouten et al15 with minor modifications. Briefly, 200–400 ng DNA was diluted with milliQ to 8 µl and heated at 98°C for five minutes (GeneAmp PCR System 9700, Applied Biosystems, Foster City, California, USA). The probe mix (1.5 μl per sample) was mixed 1:1 with a salt solution (1.5 M KCl, 300 mM Tris-HCl pH 8.5, 1 mM EDTA) and samples were heated for one minute at 95°C and incubated overnight at 60°C. Next the Ligation-65 mix (MRC Holland) was added and incubated for 15 minutes at 54°C. Ligase-65 was inactivated by heating at 98°C for one minute. The ligation products were amplified by polymerase chain reaction (PCR) using the common primer set with the 6-FAM label distributed by the supplier. Amplification products were identified and quantified by capillary electrophoresis on an ABI 3100 genetic analyser, using Genescan Analysis (version 3.7) and Genotyper software, all from Applied Biosystems. When synthetic probes were used, they were first diluted to a concentration of 1 μM. A synthetic probe mix was made of 1 μl of each diluted primer and the end volume was adjusted to 250 μl with TE buffer. For each reaction, 0.5 μl of this mix was added to 1.5 μl of the SALSA P036 kit and reactions were carried out as described above.
Mutation screening
The entire coding region of the Eu-HMTase1 gene was sequenced in 15 of the 37 selected patients using genomic DNA as a template. Primers were designed to amplify all the 25 exons of the gene as well as five alternative exons in splice variants. Primer sequences and PCR conditions are available upon request. PCR products were directly sequenced after purification with a Qiagen PCR purification kit (Qiagen Inc, Valencia, California, USA), on an Applied Biosystems 3730 automated sequencer using the same primers as in the PCR.
Expression studies
Mouse Eu-HMTase1 expression pattern
With the expressed sequence tag (EST) information contained in the NCBI unigene database, an (in silico) expression pattern of the Eu-HMTase1 gene was generated. All ESTs in the Eu-HMTase1 unigene cluster (Mm.24176), and their associated dbEST library IDs (LIDs), were re-clustered according to the tissues from which the libraries originated. A listing of the tissues used for all EST libraries was collected from the NCBI Unified Library Database (UNILIB).
Tissue in situ hybridisation
A 834 bp probe was developed by selecting a primer pair with T3 and T7 stretches (5′-taaccctcactaaagggagatgtcctaatgttcagtaatgg-3′ and 5′- taatacgactcactatagggccaagtgtcggcactcaagc-3′) that allowed the specific amplification of the 3′UTR of the mouse orthologue of Eu-HMTase1. Sense and antisense labelled cDNA probes were developed by using T7 and T3 RNA polymerase, respectively. The in situ hybridisation (ISH) was carried out according to the procedures described by Smidt et al,16 on embryos of stage E14.5 and E16.5 and on adult brain, to evaluate the expression pattern.
RESULTS
9q− phenotype in a patient with a t(X;9)(p11.2;q34.3)
The propositus is a white girl born after 37 weeks gestation, with a birth weight of 3100 g (25th centile) and a birth length of 49 cm (25th centile). Pregnancy and delivery were uneventful. She was the second daughter of healthy, non-consanguineous parents. In the neonatal period hypotonia and feeding problems were present. The anterior fontanel was enlarged and closed at the age of two years.
At two years, an atrial septal defect type II with pulmonary stenosis was diagnosed, with no haemodynamic consequences at the time.
Psychomotor development was delayed. She walked at the age of 2½ years, with hardly any speech development. She had a mild to moderate conductive hearing loss on both sides. At four years, she had epileptic seizures with EEG abnormalities, but thereafter no more seizures occurred and the EEG patterns returned to normal. She had severe behavioural problems involving autistic features, aggressiveness, and automutilation. Menarche occurred at 10 years. When we examined her at the age of 11, she was a non-cooperative, mildly obese girl with apparently normal height and head circumference, brachycephalic skull, and facial dysmorphism (synophrys, upslanting of the palpebral fissures, hypertelorism, mid-face hypoplasia, broad nasal bridge, anteverted nares, protrusion of the tongue, thickened lower lip, pointed chin, and small ears (fig 1)).

The index patient with a disruption of Eu-HMTase1 at age 11. Note brachycephaly, flattened face, upward slanting of palpebral fissures, hypertelorism, synophrys, broad nasal bridge, anteverted nares, protrusion of the tongue, thickened lower lip, and pointed chin. (Informed consent for publication of the clinical pictures has been obtained.)
Table 1 provides a summary of clinical features in 22 previously reported patients with subtelomeric 9q34.3 deletions.7–9 Stewart et al8 recently analysed the clinical and molecular characteristics of 17 of these patients. Comparing the characteristics observed in the index patient with the clinical features of the other reported patients, the overlap is striking (table 1). Features commonly seen and present in the index patient are: severe mental retardation, hypotonia, brachycephaly, mid-facial hypoplasia with synophrys and/or arched eyebrows, anteverted nares, carp shape mouth, thick lower lip, tongue protrusion, and heart defects. In addition, behavioural problems, sleep disturbances, and seizures were present in the proband as well.
Clinical features of patients with 9q34.3 submicroscopic subtelomeric deletion
Eu-HMTase1 is disrupted by the break point at chromosome 9
By previous studies, we sequenced the break points of the translocation t(X;9)(p11.2;q34.3) and found that the coding sequences of ZNF81 and Eu-HMTase1, respectively, were disrupted.12 In fig 2 (panels A and B), we show a schematic drawing of the subtelomeric region of 9q34.3 and the derivative chromosome 9 with the fusion of the disrupted genes ZNF81 and Eu-HMTase1. Translocations have been reported previously in patients with 9q− syndrome, but these were always unbalanced and associated with loss of 9qter material. However, sequence analysis of both the X chromosomal and chromosome 9 break points showed that the translocation described here is fully balanced at the genomic level (data not shown). From the der(9) chromosome, a chimeric transcript is produced consisting of part of the ZNF81 gene and an Alu-repeat sequence derived from chromosome 9. This Alu-repeat is in reverse orientation compared with the Eu-HMTase1 gene and therefore has the property of inhibiting the expression of the normal Eu-HMTase1 gene by mechanisms such as RNAi.17 To investigate this, we undertook reverse transcriptase polymerase chain reaction (RT-PCR) studies on the Eu-HMTase1 gene in lymphoblasts of the proband, but a transcript could still be detected (data not shown).

Schematic view of the subtelomeric region of 9q34.3, including the minimum critical region of 1.2 Mb (A), the derivative chromosome 9 (B), and the Eu-HMTase1 protein (C). (A) Positions and orientation of the genes MRPL41, Eu-HMTase1, and CACNA1B in the minimum 1.2 Mb critical interval. Vertical arrow heads indicate the positions of the probes used in the MLPA. (B) Schematic diagram (not to scale) indicating the exon–intron structures of ZNF81 and Eu-HMTase1 relative to the break point region. The break point in Eu-HMTase1 is situated 11 bp from the 3′ end of exon 8, resulting in a derivative chromosome 9 with a fusion of the 5′ end of the gene Eu-HMTase1 to the 5′ end of the X chromosomal gene ZNF81. The translocation was found to be fully balanced on the genomic level. (C) The positions of the four novel identified polymorphisms in the protein. Note the S809C and D229N which are present in the conserved ankyrin repeats but also identified in one of the two parents of the respective patients.
The finding of a disturbed Eu-HMTase1 in the translocation patient and the large clinical overlap of the phenotype of this patient and patients with subtelomeric 9q deletions, as shown in table1, strongly indicates that haploinsufficiency of Eu-HMTase1 is likely to be responsible for the majority of the signs and symptoms.
Tissue in situ hybridisation
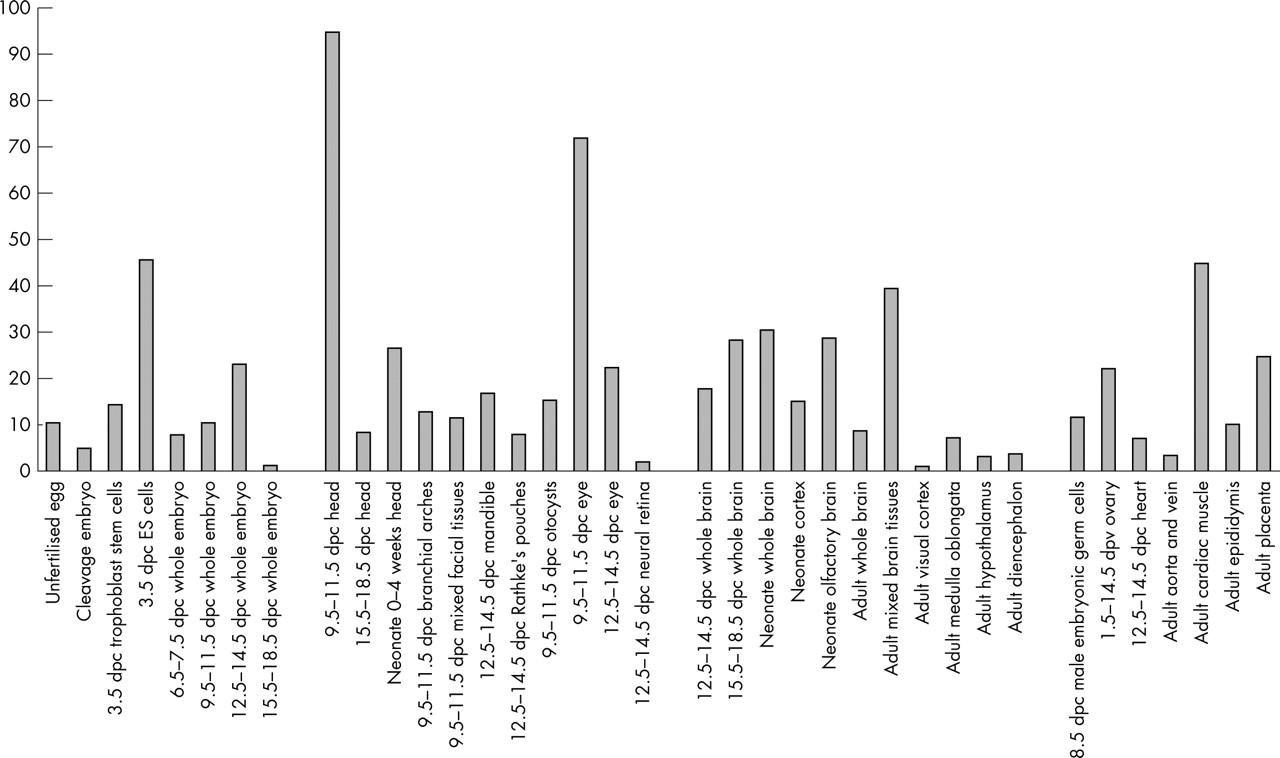
Counting the numbers of Eu-HMTase1 derived transcripts in the unigene database showed that Eu-HMTase1 is expressed throughout embryonic development, starting as early as the fertilised oocyte (fig 3). High relative expression was also observed in cDNA libraries obtained from neonatal and adult brain, eye, germ cells, and adult cardiac muscle.

Expression pattern of Eu-HMTase1 estimated by in silico analysis. The in silico Eu-HMTase1 expression pattern in selected mouse tissues generated from EST data in unigene cluster Mm.24176. Expression levels are indicated as numbers of EST sequences per 50 000 total ESTs in each tissue.
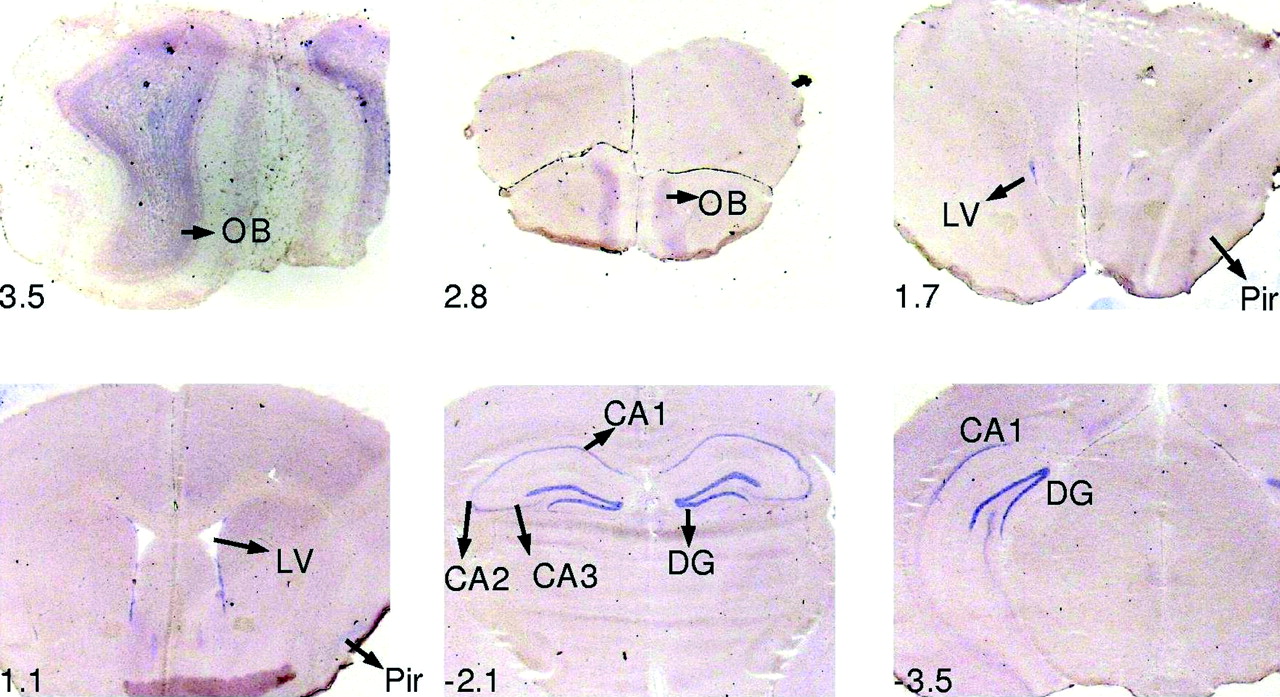
In order to obtain a more accurate impression of the spatiotemporal expression of Eu-HMTase1 during development, we designed a probe suitable for tissue ISH. This analysis revealed that Eu-HMTase1 is expressed in a broad array of structures in the developing mouse (figs 4 and 5). At these stages Eu-HMTase1 is expressed in all structures of the central nervous system (CNS). Interestingly, the developing cortex shows a laminar structure at E16.5 indicating that regulation of this gene occurs in the specific layers. The embryonic expression pattern is restricted in the adult brain to the olfactory bulb, the anterior/ventral ventricular wall, the hippocampus, and the piriform cortex (fig 6). Outside the CNS several tissues express Eu-HMTase1 (figs 4 and 5), including the developing nasal epithelium, tongue, thymus, gut, lungs, and kidney.

Eu-HMTase1 ISH on sagittal sections of a mouse embryo of stage E14.5. (A) Overview of a lateral and medial section through the embryo. (B) Lateral section showing expression in the developing retina (Ret). (C) More medial section showing expression in the lateral part of the forebrain area (CNS). (D) More medial view on the developing head. Expression was indicated in the olfactory epithelium (OF), the dorsal surface of the tongue (dT) and in the tongue itself, and again in the developing nervous system (CNS). (E) Developing body structures. Expression was clearly found in the lung and kidney. (F) Expression in the dorsal root ganglia (DRG) and again in the CNS.

Eu-HMTase1 ISH on sagittal sections of a mouse embryo of stage E16.5. (A) Overview of a lateral and medial section through the embryo. The expression in the thymus and spinal cord (SpC) is indicated. (B) Lateral section of the embryo head showing expression in the retina (Ret) and CNS. (C) Expression in the regions of the CNS including the developing hippocampus (Hip); also note the layer formation of the developing cortex. OF, olfactory epithelium; Pit, pituitary. (D) Expression in the developing whiskers (Whis) and CNS. (E) Expression in the lung, dorsal root ganglia (DRG), gut, and kidney.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In situ hybridisation on coronal sections of adult mouse brain from rostral to caudal (bregma 3.5 to −3.5) using a DIG labelled RNA probe specific for Eu-HMTase1. In rostral sections expression was detected in the olfactory bulb (OB) and the ventral ventricle wall (LV). More caudal expression was detected in different areas of the hippocampus (CA1, CA2, CA3, DG (dentate gyrus)) and weak in the piriform cortex (Pir). The position of the sections is shown relative to bregma.
MLPA analysis and mutation screening
Haploinsufficiency for the Eu-HMTase1 gene in the index patient and in patients with a 9qter deletion causes a strikingly similar phenotype. Therefore, it is likely that microdeletions or other inactivating mutations in the Eu-HMTae1 gene may account for other patients with a similar phenotype but without a demonstrable microscopic deletion. To investigate this, we selected 37 patients with a comparative phenotype for MLPA studies on subtelomeric submicroscopic 9q deletions, and subsequently carried out mutation analysis of the Eu-HMTase1 gene in 15 of them. MLPA probes were selected for the Eu-HMTase1 gene and flanking genes MRPL41 and CACNA1B (fig 2A). While MLPA analysis provided clear evidence for monosomy of chromosome 9qter in a previously reported deletion patient,6 the selected probes failed to detect any new deletions in the 37 patients whom we selected (data not shown). Direct sequence analysis of the Eu-HMTase1 gene in 15 of these patients identified eight sequence variants that were predicted to result in amino acid substitutions in the Eu-HMTase1 protein (fig 2C). Four of these were already known in SNP databases and are likely to be polymorphisms (table 2). Segregation studies of the other four changes revealed that each of the changes was present in one of the phenotypically normal parents, indicating that these represent polymorphisms as well.
Changes found with direct sequencing in the Eu-HMTase1 gene
DISCUSSION
We here describe how a patient with a disruption of Eu-HMTase1 has the same clinical characteristics as are seen in patients with a submicroscopic subtelomeric 9q deletion (9q−). This strongly indicates that haploinsufficiency of Eu-HMTase1 is responsible for the phenotype in this patient as well as for the major part of the 9q− syndrome. Further support for this is provided by a parallel paper in which two patients are reported with a 9q− phenotype and microdeletions that define the critical region encompassing two genes including Eu-HMTase1.18 Our results provided a rationale for mutation screening in patients with a similar phenotype. We carried out MPLA in 37 patients sharing some of the clinical features and sequenced the entire coding region in 15 of them. In none of these patients were Eu-HMTase1 abnormalities detected. However, this does not imply that such patients do not exist. The numbers tested here may have been too small to identify additional mutations, and patients with a phenotype similar to that seen in individuals with 9q− syndrome but without chromosomal abnormalities are scarce. To increase the chances of identifying such deletions, we have included the reported Eu-HMTase1 probes in the standard MLPA kit that is used for the detection of subtelomeric deletions in mentally retarded patients in our DNA diagnostic services.
The phenotypic overlap between the patient with the disrupted Eu-HMTase1 gene and the 9q− syndrome is striking (fig 1, table 1). All the major features of the 9q− syndrome were present in the translocation patient, in addition to some less reported features: behavioural problems, sleep disturbances, and seizures. The only significant exception is microcephaly, which is common in 9q− patients (18 of 22 cases), but absent in the translocation patient. This may just be a reflection of the clinical variability that characterises 9q− syndrome patients. Another likely explanation is that the microcephaly in 9q− syndrome is not caused by haploinsufficiency for the Eu-HMTase1 gene, but by deletion of another gene from the subtelomeric region of chromosome 9q. This explanation may also apply to some of the other less frequent clinical features such as soft brittle hair with partial alopecia and urinary tract anomalies.
According to EST data extracted from electronic databases, Eu-HMTase1 is abundantly expressed in embryonic stem cells, in brain and eye during embryonic development, in various adult brain tissues, and in adult cardiac muscle. We have shown using ISH that Eu-HMTase1 is expressed in a broad array of structures in the developing mouse, whereas in the adult mouse, the expression pattern is confined to a few regions of the brain. This expression pattern of the Eu-HMTase1 supports the hypothesis that this gene is an excellent candidate as a cause of mental retardation and congenital abnormalities. Expression in the adult brain is restricted to specific areas of the olfactory bulb, the anterior/ventral ventricular wall, the hippocampus, and the piriform cortex. Interestingly, these structures are all known to harbour cell populations with high proliferative potential.19–21 As widespread expression of Eu-HMTase1 is also observed in embryonic CNS development, it appears that this gene has an important role in proliferation of neuronal precursor cells in neurogenesis. Alternatively, the high expression of the gene in regions containing dividing cells may also point to a role of the Eu-HMTase1 protein in mitotic silencing of neuronal precursor cells after cell division. For both of these postulated functions it is of interest that Eu-HMTase1 acts as an epigenetic regulator of transcription.
Eu-HMTase1 was identified in 200222 and was found to be closely related to the G9a protein. Both Eu-HMTase1 and G9a are H3-K9 HMTases which are present in euchromatic regions and they form complexes with heterochromatin protein 1 (HP1γ) and a subset of E2F transcription factors.22 The E2F-6 complex preferentially occupies target promoters in G0 cells rather than G1 cells, suggesting a role in silencing of target genes in quiescent cells. In view of the restricted expression in adult mouse brain it appears likely that Eu-HMTase1 plays an important role in carrying out the epigenetic histone modifications which are needed to transfer a subset of the neuronal precursor cells into the G0 phase after cell division. It was shown recently that homozygous mutant mouse embryos with a mutation in G9a die at 8.5 dpc (days post-conception), implying that euchromatin H3-K9 methylation is essential for early embryogenesis.23 The findings with ESET, another H3-K9 HMTase, were similar24; these investigators reported that ESET-null embryos show peri-implantation lethality. In addition to histone methylation, H3-K9 HMTases also influence DNA methylation.25,26 Thus G9a-null embryo stem cells (ES) show altered DNA methylation in the Prader–Willi imprinted region.25 Knockout mice for Eu-HMTase1 have not been reported to date.
Studies in recent years have revealed that histone methylation plays an important role in modulating chromatin structure and function.27,28 Histone methylation occurs on arginine and lysine residues at the N-terminal tails of histones H3 and H4 and is catalysed by two distinct protein families, the PRMT1 and the SET domain containing family. Recently, a third family of histone methyl transferases (HMTases) that target lysine 79 was reported, located in the globular domain of histone H3.29 Thus far, six lysine residues located on histones H3 (lysines 4, 9, 27, 36, and 79) and H4 (lysine 20) have been reported to be sites of methylation. Lysine 9 methylation (H3-K9) is the best studied owing to its fundamental role in chromatin formation, transcriptional silencing, X chromosome inactivation. and DNA methylation.30
There is a growing number of mental retardation genes that appear to play a role in chromatin remodelling: MECP2, RSK2, XNP, DNTM3B, and CBP.31 Three of these genes—MECP2, RSK2, and XNP—are involved in syndromic and non-syndromic forms of X linked mental retardation (XLMR). Mutations in the MECP2 gene cause RTT syndrome in females, mostly in isolated cases.31,32 In addition, there are several families in which MECP2 mutations cause male limited non-specific XLMR.33,34 The MeCP2 protein is a methyl-CpG-binding protein that forms transcription repression complexes with Sin3a and several histone deacetylases.31,35,36 The RSK2 gene is involved in Coffin–Lowry syndrome37 and, in one family, in non-specific mental retardation.38 The corresponding protein is a ribosomal S6 kinase involved in RAS-MAPK-ERK signalling. In addition, it has been shown that RSK2 can phosphorylate histone H3 and the CREB binding protein CBP, which in turn acetylates histone H3.39,40 Mutations in the CBP gene are causative for Rubinstein–Taybi syndrome.41 The XNP gene is the third X chromosomal XLMR gene with a role in chromatin remodelling. Mutations in the XNP gene cause the ATRX syndrome and various other syndromes with overlapping clinical features.42 Also, non-specific XLMR has been reported in two families.43,44 XNP (X linked nuclear protein) is a member of the Swi/Snf family of RNA helicases. Interaction between the XNP protein and two heterochromatin binding proteins, HP1 and EZH2, is well documented.45 Interestingly, two recently identified XLMR genes also encode predicted interactors of HP1, KRAB-ZNF (Krüppel associated box-zinc finger proteins) and the related ZNF41 and ZNF81.12,46
The current identification of Eu-HMTase1 as the causative gene in yet another mental retardation syndrome strongly suggests that disruption of epigenetic patterns is a common cause of mental retardation and indicates that this type of transcriptional regulation is important in normal brain development.
ELECTRONIC DATABASE INFORMATION
Accession numbers and URLs for data presented in this report are as follows:
-
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for Eu-HMTase1 accession number NM_ Q9H9B1 (human) and NM_BC056938 (mouse).
-
MRC-Holland, http://www.mrc-holland.com/ (for SALSA P036 Human Telomere Test Kit) MLPA, http://www.mlpa.com/mlpa_protocols.htm
-
UCSC Genome Browser (Golden Path), http://genome.ucsc.edu/cgi-bin/ (July 2003 and May 2004 Build)
-
NCBI Unified Library Database (UNILIB), http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi
Acknowledgments
This work was supported by grants from ZonMw, grant 940-37-031 (TK) and ZonMw, grant 907-00-058 (BdV), Dutch Brain Foundation, grants 12F04(27)64 and 10F02(2)43 (HY), and an EU grant, QLG3-CT-2002-01810 (HvB; EURO-MRX).
REFERENCES
Footnotes
-
Competing interests: none declared