Article Text

Abstract
Introduction: Mutations in the genes encoding collagen VI (COL6A1, COL6A2, and COL6A3) cause Bethlem myopathy (BM) and Ullrich congenital muscular dystrophy (UCMD). BM is a relatively mild dominantly inherited disorder with proximal weakness and distal joint contractures. UCMD is an autosomal recessive condition causing severe muscle weakness with proximal joint contractures and distal hyperlaxity.
Methods: We developed a method for rapid direct sequence analysis of all 107 coding exons of the COL6 genes using single condition amplification/internal primer (SCAIP) sequencing. We have sequenced all three COL6 genes from genomic DNA in 79 patients with UCMD or BM.
Results: We found putative mutations in one of the COL6 genes in 62% of patients. This more than doubles the number of identified COL6 mutations. Most of these changes are consistent with straightforward autosomal dominant or recessive inheritance. However, some patients showed changes in more than one of the COL6 genes, and our results suggest that some UCMD patients may have dominantly acting mutations rather than recessive disease.
Discussion: Our findings may explain some or all of the cases of UCMD that are unlinked to the COL6 loci under a recessive model. The large number of single nucleotide polymorphisms which we generated in the course of this work may be of importance in determining the major phenotypic variability seen in this group of disorders.
- BM, Bethlem myopathy
- PTC, premature termination codon
- SCAIP, single condition amplification/internal primer
- UCMD, Ullrich congenital muscular dystrophy
- vWF, von Willebrand factor
- Bethlem myopathy
- collagen VI
- genomic sequencing
- Ullrich congenital muscular dystrophy
Statistics from Altmetric.com
- BM, Bethlem myopathy
- PTC, premature termination codon
- SCAIP, single condition amplification/internal primer
- UCMD, Ullrich congenital muscular dystrophy
- vWF, von Willebrand factor
Extracellular matrix molecules are critical for skeletal muscle stability, regeneration, and muscle cell matrix adhesion.1–3 Collagen VI is a ubiquitous extracellular matrix protein4 which forms a microfibrillar network in close association with the basement membrane around muscle cells and which is capable of interacting with several other matrix constituents.5–7 Collagen VI is composed of three different peptide chains: α1(VI) and α2(VI), both 140 kDa in size, and α3(VI) which is much larger at 260–300 kDa.8 The α1(VI) and α2(VI) chains are encoded by two genes (COL6A1 and COL6A2, respectively) situated in a head-to-tail organisation on chromosome 21q22.39 (NT_011515) and separated by 150 kb of genomic DNA, whereas COL6A3, the gene for the α3(VI) chain, maps to chromosome 2q3710 (NT_005120). All three chains contain a central short triple helical domain of 335–336 amino acids with repeating Gly-Xaa-Yaa sequences flanked by large N- and C-terminal globular domains consisting of motifs of 200 amino acids each homologous to von Willebrand factor (vWF) type A domains.11 In total, the three COL6 genes consist of 107 coding exons spread over 150 000 bp of genomic DNA. COL6A2 and COL6A3 undergo extensive alternative splicing.12–14
The assembly of collagen VI is a complex multistep process. Association of the three subunits, α1(VI), α2(VI), and α3(VI), to form a triple helical monomer is followed by staggered assembly into disulfide bonded antiparallel dimers, which then align to form tetramers, also stabilised by disulfide bonds. Outside the cell, tetramers, the secreted form of collagen VI, associate end-to-end to form the characteristic beaded microfibrils.8,15,16
Mutations in the genes encoding collagen VI cause Bethlem myopathy (MIM 158810) and Ullrich congenital muscular dystrophy (MIM 254090), two increasingly recognised conditions which were previously believed to be completely separate entities. Bethlem myopathy (BM) is a dominantly inherited disorder which usually follows a relatively mild course and is characterised by proximal muscle weakness and joint contractures mainly involving the elbows, ankles, and fingers.17,18 Where contractures are prominent, this disorder may resemble an Emery-Dreifuss muscular dystrophy.19 In other patients the contractures may be subtle, leading to confusion in diagnosis with cases of limb-girdle muscular dystrophy.20 By contrast, Ullrich congenital muscular dystrophy (UCMD) is an autosomal recessive condition causing severe muscle weakness of early onset such that children may never walk independently, with proximal joint contractures and striking hyperelasticity of distal joints, and early respiratory failure.21–23
To date, 11 different dominant mutations in the COL6 genes have been published in BM patients. Truncating and splicing mutations in COL6A1 affecting protein folding and secretion as well as a missense mutation in the COL6A3 N-terminal α3(VI) domain are thought to cause BM via functional haploinsufficiency.24–28 In other cases single amino acid substitutions disrupting the Gly-Xaa-Yaa motif of the highly conserved triple helical domain of any of the three collagen VI chains20,29,30 appear to interfere with extracellular microfibril formation. Ten recessive truncating mutations in COL6A2 and COL6A3 have been described in eight families with UCMD, eight in COL6A2,31–34 and two in COL6A3.35 The mild phenotype of one of the latter, where a homozygous nonsense mutation leads to a stop codon in exon 5 of COL6A3, can possibly be explained by alternative splicing. In addition, a homozygous splice site mutation causing an in-frame deletion of 17 amino acids of the triple helical region of the α3(VI) chain has been associated with a comparatively mild phenotype.35 Recently, there has also been one report of an apparently dominant de novo heterozygous in-frame deletion of exons 9 and 10 of the α1(VI) chain in a patient with a classical UCMD phenotype.36
In the patients with UCMD and COL6 mutations reported so far, immunohistochemical analysis of muscle biopsy samples shows absent or severely reduced collagen VI expression especially in the basement membrane, while in most patients with BM collagen VI immunolabelling is normal (though a secondary reduction in laminin β1 has been reported in some patients). A few patients have also been recently described where sarcolemmal collagen VI expression is abnormal but mutations in the COL6 genes have been excluded by RT PCR and cDNA sequencing.32 Because of the complexities of genetic counselling caused by the possibility of dominant as well as recessive mutations in the collagen VI genes, and the possibility that collagen VI abnormalities may also occur as a secondary phenomenon, accurate molecular diagnosis becomes very important. As there are currently no other techniques available for routine molecular diagnosis in this group of diseases, a rate limiting step in diagnosis is the ability to offer mutation detection.
We have developed a method for rapid, robust, and economical direct sequence analysis of all 107 exons of the COL6 genes using single condition amplification/internal primer (SCAIP) sequencing. SCAIP relies on amplification of a large number of exons under a single set of PCR temperatures. Sufficient sequencing specificity is obtained by uniform use of a second, internal set of sequencing primers without optimisation of individual amplicon conditions.
SCAIP was developed for direct sequence analysis of the dystrophin gene37 and avoids intermediate screening steps such as single-stranded conformation polymorphism (SSCP) or denaturing high-performance liquid chromatography (DHPLC). It is now used for the routine molecular diagnosis of dystrophinopathies. This is the first study to perform genomic sequence analysis of all three COL6 genes. The three COL6 genes were sequenced from genomic DNA in 79 patients with UCMD or BM, achieving over 97% coverage. Potential mutations were found in 62% of patients, thereby more than doubling the number of previously identified COL6 mutations. Most of these putative mutations are consistent with straightforward autosomal dominant or recessive inheritance, though our results suggest that a proportion of UCMD patients may have dominant mutations rather than recessive disease. In addition, we have discovered in several UCMD patients examples of putative recessive mutations in each of two different COL6A genes, potentially indicating a novel mode of disease causation or modification in this group.
METHODS
Patients
We ascertained 79 patients from four muscle centres with clinical phenotypes suggestive of either UCMD (see Pepe et al19 for diagnostic criteria) or BM (see Pepe et al38 for diagnostic criteria) and also included patients with overlapping/intermediate phenotypes in the BM cohort who had features of both connective tissue as well as skeletal muscle involvement. Of the patients, 26 were UCMD patients and 53 BM, of which nine were sub-characterised as “mild BM” and nine as “severe BM”. Their genomic DNA was extracted from peripheral lymphocytes or dermal fibroblast cultures by standard methods. The genetic studies were performed in accordance with the ethical procedures of the participating institutions. The mutations in individuals #71 (harbouring a heterozygous splice donor change at COL6A1 intron 14+1) and #72 (carrying a heterozygous internal COL6A1 gene deletion which removes exons 9 and 10) have previously been published36 and were used as positive controls.
SCAIP sequencing
The genomic organisation of the COL6 genes was assembled from contigs downloaded from the Nov2002 assembly of the UCSC Human Genome Browser web page39 (http://genome.cse.ucsc.edu/; see also the International Human Genome Sequencing Consortium 200140). Assembly and exon-intron annotation was performed using task specific Perl scripts. Our assembly reveals that the collagen VI regions on chromosome 21q22.3 and 2q37 are currently contiguous and gap free for the COL6A1 transcript (accession number NM_001848), the COL6A2 isoforms 2c2 (accession number NM_001849), 2c2a (accession number NM_058174), and 2c2a′ (NM_058175) as well as the full length isoform of COL6A3 (accession number NM_004369) and its four variant transcripts (NM_057164, NM_057165, NM_057166, and NM_057167). Primer systems for PCR were designed to span each coding exon including splice variants. Each amplicon was designed for an optimal size range of 1.2–1.4 kb with the exon centred within the amplicon to maintain uniform conditions. This resulted in 103 amplicons of nearly uniform size with the uniformity allowing prediction of likely amplification conditions using a single set of PCR temperatures. Aliquots (15 pmol) of each primer were placed into individual wells of a 96 well plate, evaporated to dryness, and stored at −20°C until use. For PCR amplification, 25 μl of a master PCR mixture were aliquoted into each well of a thawed plate so that each well contained 100 ng of genomic DNA, 200 μM dNTPs, 15 pmol of each PCR primer, 1×reaction buffer, 2 mM Mg2SO4, 1 M GC-RICH resolution solution (Roche, Indianapolis, IN, USA), and 0.5 U Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA, USA). The PCR conditions were 2 min at 94°C; 10 cycles of 94°C for 10 s, 55°C for 30 s, and 68°C for 2 min; 20 cycles of 94°C for 10 s, 55°C for 30 s, and 68°C for 2 min, elongated by 20 s/cycle. The PCR products were then bound to a 96 well filter plate with a 1.0 μm glass fibre type B filter (Millipore, Billerica, MA, USA) in the presence of 5 M guanidine HCl/1 M potassium acetate solution and washed with 80% ethanol followed by elution with warm nanopure H2O.
Internal sequencing primers were designed and stored in 384 well plates so that both PCR set up and sequence reaction set up could be done with multichannel pipettors and pipetting robots. For sequencing 6 μl of PCR product and 10 pmol of sequencing primer were evaporated to dryness in a speed vacuum system and then rehydrated with 0.5 μl ABI Prism BigDye terminators (version 3.0) and 0.75 µl 5×buffer mix, and made up to 5 μl with nanopure H2O before the plates were heat sealed with foil. The sequencing conditions were 30 s at 96°C, 46 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min following which the sequencing reactions were precipitated with 20 μl of 62.5% ethanol/1 M potassium acetate (pH 4.5) and centrifuged at 4000 rpm at 4°C for 45 min. Following resuspension in 15 μl HiDi formamide (ABI; Applied Biosystems, Foster City, CA, USA) the samples were electrophoresed on an ABI 3700 DNA analyser prepared with POP-5 capillary gel matrix.
The oligonucleotide sequences of primers used are available in supplementary table A (only available online at http:/jmg.bmjjournals.com/supplemental/).
Sequence analysis
Following initial data processing on the ABI 3700 instruments, the base calls were analysed with the Phred programme41 which adds a quantitative base quality value and provides a probability of the correctness of a base call. The quality values represent the log of the probability that the base call is correct, such that a Phred value of 20 corresponds to a 99% probability of accuracy and a Phred value of 30 corresponds to a 99.9% probability that a base call is accurate. Using the Phrap programme42 the read sequence was assembled with consensus sequence and potential mutations were identified using the Consed programme.43 The read assembly was done on a per-PCR fragment basis and a single Phrap assembly consisted of the consensus genomic sequence and all sequence reads relating to the PCR fragment. The read sequence and Phred quality values were compared with the assembled consensus sequence using Cross_match,42 and all discrepancies were tagged and ranked according to Phred quality of the base. Potential base discrepancies were catalogued using Perl scripts and their trace files underwent human review. The two control samples were analysed in a blinded fashion.
Significance of sequence changes
We analysed all three genes in 158 chromosomes, thereby allowing internal controls for the possibility that sequence changes might represent polymorphisms. We adopted a conservative approach to the designation of sequence changes as possibly pathogenic. Sequences showing variation from the published data were assigned as likely to be polymorphisms if they were at intron positions ±4 or more nucleotides away from the splice site or synonymous exonic changes. In addition, exonic variants leading to non-synonymous amino acid changes were designated as polymorphisms if they had been reported previously, were present as an additional change in patients with already defined mutations, were present in patients known to be unlinked to the particular gene in question, or the minor allele had been annotated as the consensus in the GenBank reference sequence. Conversely, sequence variations were designated as putative mutations if they were rare or unique exonic changes leading to non-synonymous amino acid changes, splice acceptor or splice donor changes at position ±1–3 or changes of the last nucleotide in the exon, or exonic insertions or deletions leading to shift of the reading frame and a consequent premature termination codon, or involving a splice site.
Sequence changes assigned as putative mutations were resequenced from a second PCR product to confirm the reliability of the original finding.
RESULTS
Details of all the putative mutations and polymorphisms found are stratified by gene in tables 1–3. The patient specific putative mutations are documented in table 4 and in fig 1.
Putative mutations and probable polymorphisms found in COL6A1
Putative mutations and probable polymorphisms found in COL6A2
Putative mutations and probable polymorphisms found in COL6A3
Putative COL6 mutations for each patient

{kind=link}
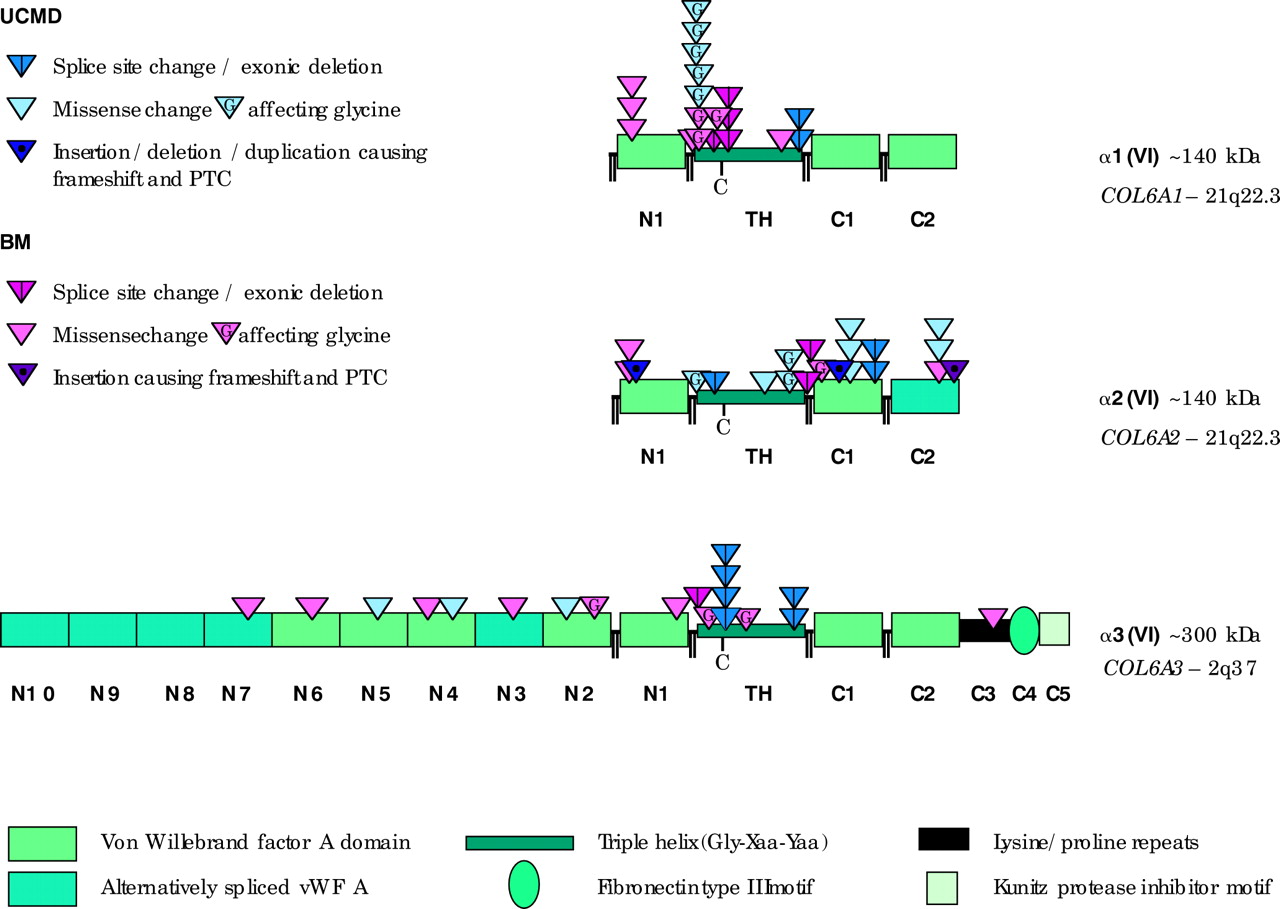
Localisation of the identified putative mutations. Schematic diagram of the domain structure of the protein chains encoded by COL6A1-3. All three chains contain a triple helical domain of 335–336 amino acids flanked by large N- and C-terminal globular domains made up of vWF A-like motifs. Short cysteine rich connecting peptides separate N2, N1, triple helix, C1, and C2. The triple helical domains contain a single cysteine residue (depicted as “C”) which is important for dimer assembly. The localisation of the identified putative mutations is shown stratified by clinical phenotype.
BM
Our BM cohort of 53 patients included three affected siblings (two of whom had milder symptoms than the third), as well as four parent-offspring pairs. Counting each family only once, we found 23 different putative mutations in 26 of 47 patients with a clinical diagnosis of BM. Seven are in COL6A1 (one found in two unrelated individuals and two each found in three unrelated individuals), six in COL6A2 (one recurring in two unrelated individuals), and 10 in COL6A3. Of these, five are predicted to alter splicing (two in COL6A1, two in COL6A2, and one in COL6A3), one in COL6A2 is predicted to lead to frameshift and a consequent premature termination codon, and 17 are predicted to cause non-synonymous amino acid substitutions (five in COL6A1, three in COL6A2, and nine in COL6A3) and are thus putative missense mutations.
One of the putative missense mutations, a G to A change in exon 11 of COL6A3, which is predicted to change a glycine to glutamic acid in the N-terminal domain of the α3(VI) chain, was previously reported by Pan et al.25 Another four are predicted to cause single amino acid substitutions disrupting the Gly-Xaa-Yaa motif of the highly conserved triple helical domain of any of the three COL6 genes.
The three affected siblings, #3, #4, and #9 in family ε, share a heterozygous G to A change in exon 26 of COL6A2 which is predicted to change a glycine to serine in the C1-terminal domain of the α2(VI) chain. Two separate putative missense mutations, one in COL6A1 and one in COL6A3, are present in both individuals (#28 and #29) of parent-offspring pair β. The heterozygous C to T change in exon 9 of COL6A1 is predicted to change a proline to leucine in the triple helical domain of the α1(VI) chain and the heterozygous G to A change in exon 15 of COL6A3 is predicted to change a glycine to aspartic acid right at the beginning of the triple helical domain of the α3(VI) chain. Two unrelated individuals (#43 and #74) both carry one putative missense mutation in COL6A1 alongside a putative missense mutation in COL6A3. We also found a splice donor mutation in COL6A1 intron 14+1 (first described by Lamande et al44) in our parent-offspring pair γ and two other unrelated individuals, one of the latter being positive control individual #71 who has previously been reported on by Pan et al.36
UCMD
In the 26 patients with a UCMD phenotype, a total of 21 different putative mutations were found in 20 unrelated patients. Four are in COL6A1 (two of which are present in two unrelated individuals), 11 are in COL6A2, and six in COL6A3 (one of which is present in three unrelated individuals). Of these, seven are predicted to alter splicing (one in COL6A1, three in COL6A2, and three in COL6A3), two are predicted to lead to premature termination codons in COL6A2, and 12 are predicted to cause non-synonymous amino acid substitutions (three in COL6A1, six in COL6A2, and three in COL6A3) and are thus putative missense mutations. Single amino acid substitutions disrupting the triple helical Gly-Xaa-Yaa motif are predicted to be caused by five of these changes. Within the UCMD cohort, five patients are homozygous for putative mutations and two patients have two different changes in the same gene, whereas in another 10 patients only one putative mutation was identified (five of these are predicted to result in a splicing error and five are putative missense mutations). Finally, two patients are compound heterozygotes with one putative mutation each in COL6A1 and COL6A2.
We also documented 145 polymorphisms (34 in COL6A1, 49 in COL6A2, and 62 in COL6A3). Of these, 68 are intronic (24 in COL6A1, 30 in COL6A2, and 14 in COL6A3), 50 are exonic but predicted to be synonymous (seven in COL6A1, 11 in COL6A2, and 32 in COL6A3), and 27 are exonic and predicted to cause a non-synonymous amino acid substitution (three in COL6A1, eight in COL6A2, and 16 in COL6A3). Of the latter, nine had been reported previously,36 four were present in a patient known to be unlinked to the particular gene in question, three were registered in the NCBI dbSNP database, three were designated as polymorphisms following personal communication with Shireen Lamande (Cell and Matrix Biology Research Unit, University of Melbourne, Australia, 2004) regarding unpublished polymorphism data, six were present as additional changes in patients with an already defined mutation, one was present in only one of two affected first degree relatives in a pedigree with autosomal dominant inheritance, and for one the minor allele had been annotated as the consensus in the GenBank reference sequence.
In addition, our genomic sequencing data revealed that the COL6A1 GenBank transcript NM_001848 harbours two inaccuracies. At position c.1182_1185 there are only three G present instead of the four found in the GenBank sequence. Similarly, at position c.1192_1193 there are three G instead of the two found in GenBank. These results alter the predicted amino acid sequence: assuming an exon 17 splice acceptor sequence of CAG, we predict that the amino acids number 396 and 397 within the triple helix of the α1(VI) chain would be glutamine and proline instead of proline and alanine within the Gly-Xaa-Yaa motif.
DISCUSSION
SCAIP has previously been demonstrated to be efficient in detecting mutations in the X linked gene dystrophin.37 We have now applied it to the three COL6 genes and shown that it is a reliable method in these autosomal genes. We have sequenced all three COL6 genes from genomic DNA in a cohort of 79 patients clinically ascertained as either UCMD or BM with more than 97% coverage of the 107 coding exons for all patients. We found putative mutations in one of the COL6 genes in more than 62% of patients (66% of those clinically classified as BM, 56% of severe BM patients, and 79% of UCMD patients). The comparatively low detection rate in patients with mild BM (22%) and patients with UCMD and short stature (0%) may be related to overlap of their phenotype with other forms of muscular dystrophy.
Sequence changes detected
The conservative approach we adopted to the designation of sequence changes as possibly pathogenic was guided by the criteria suggested by Cotton and Scriver.45 In the clinical context a “mutation” is generally regarded as a genetic variant which has a harmful effect and causes disease. Our project focussed on genomic sequencing and did not extend to segregation or expression studies, work which will be necessary to confirm the pathogenicity of sequence variants we have detected but which was outside the scope of our project. We thus designated variants as putative mutations by using criteria of mutation type.
Variants likely to affect splicing
We found nine different sequence variations at intron position ±1–3 from the splice site as well as one complex insertion/deletion involving a splice donor site and one deletion involving a splice acceptor site. These change the highly conserved “sequence logos” of the spliceosome described by Stephens and Schneider.46 We also included a homozygous change of the last exonic nucleotide adjacent to the COL6A3 exon 27 splice donor site in a patient from a consanguineous relationship known to link to COL6A3 as a putative splice mutation.47 In order to confirm the pathogenicity of all putative splice mutations it will be necessary to perform RT PCR on RNA from dermal fibroblasts of these patients. Seven of the splice site changes were in UCMD and five in BM patients.
Variants likely to cause functional haploinsufficiency
One exonic variant directly generates a premature termination codon in UCMD patient #78 and two duplications are predicted to change the reading frame creating downstream premature stop codons following six or nine non-synonymous amino acids, in BM patient #70 and UCMD patient #23, respectively. These changes would be expected to lead to premature mRNA decay and subsequent haploinsuffiency.46
Likely pathogenic missense changes
For the sequence variations predicted to cause non-synonymous amino acid substitutions, the differentiation between putative missense mutation and probable polymorphism is more difficult. A polymorphic variation is without phenotypic effect and is formally defined as a change present at a frequency of more than 1% of alleles in a population. Given the absence of normal controls in our cohort, we did not deem it justified to assign every change occurring with a frequency of more than 1% as a probable polymorphism, especially because certain sequence variations conforming to a well described pathogenic mechanism—the disruption of the Gly-Xaa-Yaa motif of the highly conserved triple helical domain20,29,30—occurred with a frequency higher than that. For example, a heterozygous G to A change in exon 10 of COL6A1, predicted to change a glycine to arginine within the triple helical domain of the α1(VI) chain, occurred in 4/77 unrelated individuals, that is, with an allele frequency of 2.6%. Conversely, we noticed that some previously published polymorphisms, such as the G→A change in exon 18 of COL6A2 thought to substitute a glutamine for arginine, occurred with extremely low frequency (in this case 1/156 chromosomes, that is, 0.6%) in our cohort.
We identified 55 different exonic sequence variations that are predicted to cause non-synonymous amino acid substitutions. Analysing their frequency, we noticed that all variants fell into one of two categories, occurring either with an allele frequency of more than 20% or with a frequency of less than 3%. The former, which had all been previously reported or were registered in the NCBI dbSNP database, were designated as probable polymorphisms. Within the latter group we assigned putative missense mutation status to all changes, unless they had been previously reported or registered, were present as an additional change in a patient with a proven mutation, were present in a patient known to be unlinked to the particular gene, or only found in one of two affected first degree relatives of a family with an autosomal dominant pedigree.
Using these criteria, 28 of the 55 identified exonic sequence variations predicted to cause non-synonymous amino acid substitutions are considered to be putative missense mutations (11 found in UCMD patients, 16 in BM, and one in both UCMD and BM).
The pathogenic mechanism of single amino acid substitutions disrupting the Gly-Xaa-Yaa motif of the highly conserved triple helical domain of any of the three collagen VI chains has been previously described20,29,30 and this is likely to apply to nine of our 28 changes which are distributed in equal numbers between BM and UCMD patients. One of these, a G to T change in exon 14 of COL6A1 which is predicted to change a glycine to valine, corresponds to a glycine to aspartic acid change at this position and has previously been reported.20 Another variant, a G to A change in exon 11 of COL6A3 which is predicted to change a glycine to glutamic acid in the N-terminal domain of the α3(VI) chain, was previously reported by Pan et al.25 Although other missense mutations not concerning glycines in the triple helical domain have been published,20 for the remainder of our putative missense mutations, further expression studies as well as segregation analysis and testing of large numbers of healthy controls will be necessary to confirm their pathogenicity or reassign them as polymorphisms.
UCMD
Within the UCMD cohort, five patients were found to be homozygous for putative mutations and two patients have two different sequence changes in the same gene which would be in keeping with the autosomal recessive mode of inheritance originally described for this condition. However, in another 10 UCMD patients only one putative mutation was identified. While it is possible that these patients might carry a second mutation not detectable by our technique, such as a large exonic deletion, an alternative possibility is that the sequence variations identified in these patients might act in a dominant fashion, similar to the mutation reported by Pan et al.36 If this is confirmed, dominant mutations may be a common finding in patients with features suggestive of UCMD. This could explain the apparent lack of linkage to COL6A genes in patients with a UCMD phenotype according to a recessive model. So could the finding of two patients with a clinical UCMD phenotype (#45 and #48) who are compound heterozygotes for putative mutations in COL6A1 and COL6A2.48 Both carry a sequence variation predicted to cause a glycine substitution within the triple helical domain of the α1(VI) chain alongside a separate unique putative missense mutation in COL6A2. These glycine substitutions may act in a dominant fashion or may be present together with an undetected large exonic deletion. However, given the potential pathogenicity associated with both variants, an important alternative possibility is inheritance of alterations in two different COL6 genes causing disease. Family studies for inheritance of the changes and presence or absence of the phenotype in carriers as well as expression studies will be necessary to determine whether one or both changes for either of these patients are disease causing or disease modifying. Neither of these two families have a positive dominant family history of weakness.
BM
Two separate putative missense mutations, one in COL6A1 and one in COL6A3, are also present in one of our BM parent-offspring pairs. Given that it is predicted to change a glycine to aspartic acid right at the beginning of the triple helical domain of the α3(VI) chain, the heterozygous G to A change in exon 15 of COL6A3 is likely to be disease causing. However, an additional heterozygous C to T change was also detected in this family and is predicted to change a proline to leucine in the triple helical domain of the α1(VI) chain. This change is not present in the other 77 individuals tested and could therefore be either be a rare polymorphism or convey additional pathogenicity. Similar reasoning can also be applied to the case of another unrelated BM patient, who carries a COL6A1 sequence variation predicted to change a glycine in the triple helical domain of the α1(VI) chain to arginine alongside a putative missense mutation in COL6A3, and a further BM patient where two putative missense mutations in two different COL6 genes have been identified. As discussed before, further expression studies as well as segregation analysis and testing of large numbers of healthy controls will be necessary to confirm their pathogenicity or possible role as modifiers or to reassign them as polymorphisms.
Recurrent putative mutations
Within our BM cohort of patients, a parent-offspring pair and two further unrelated individuals (one of the latter being positive control individual #71 who has previously been reported36) carry an identical splice donor mutation in COL6A1 intron 14+1 which was first described as a BM mutation by Lamande et al.44 A heterozygous exon 16 splice donor change was identified as the only putative mutation in three unrelated individuals of our UCMD cohort of patients.
Mutations found in both BM and UCMD
The putative missense mutation predicted to cause a substitution of glycine by arginine in the triple helical region of COL6A1 is present as a heterozygous change in two unrelated individuals assigned to the BM cohort. The same mutation was detected in two unrelated individuals assigned to the UCMD cohort. These two BM patients had a more severe phenotype than average, while the clinical phenotype of the two UCMD patients was comparatively mild. This underlines our impression that there is in fact a spectrum of collagen VI related disorders rather than two strictly separate entities, but further work will be necessary to delineate the exact pathogenic mechanisms and correlate these to the clinical and pathological findings.
Probable polymorphisms
Overall our results confirm that the three COL6 genes are highly polymorphic. We identified a high number of intronic and exonic variants designated to be probable polymorphisms. Of the 77 non-pathogenic exonic sequence changes we have observed in the three COL6 genes, 55 resulted in an amino acid substitution. In theory, it is possible that some of the 23 rare or unique exonic synonymous changes as well as some of the 24 rare intronic changes which we classified as probable polymorphisms might influence splicing. In patients with no immediately convincing pathogenic mutation, it will thus be important to perform RT PCR studies even for these changes.
SUMMARY
The genetics of the collagen VI related muscular dystrophies/myopathies are complex. Collagen VI is composed of three different peptide chains which are encoded by three large genes and the assembly of collagen VI involves a number of different stages. Different mutations have been shown to have variable effects on protein assembly, secretion, and its ability to form a functioning extracellular network. The multitude of possible mutational mechanisms and modes of inheritance has important consequences for the genetic counselling of patients and their families.
Our results suggest that mutations in the COL6 genes are likely to be found in the majority of patients with a clinical diagnosis of UCMD and BM, but that stringent criteria need to be applied to the designation of likely pathogenic changes in these genes because of their highly polymorphic nature. For many sequence changes demonstration of pathogenicity by segregation analysis or RT-PCR and cDNA sequencing will be necessary.
This study reports the first application of SCAIP methodology to an autosomal gene locus, and demonstrates that one of the limitations of this method for analysis of autosomal genes is the inability to detect reliably large exonic deletions, as illustrated by the fact that we were unable to detect the large heterozygous COL6A1 gene deletion known to be present in positive control individual #72. However, the intronic sequences co-amplified as part of our PCR strategy provide a rich source of polymorphisms, which when heterozygously present can help to exclude the possibility of large deletions. In addition, although SCAIP analysis provides sequence information on exon/intron junctions and on the immediately adjacent intronic sequence, it cannot detect mutations located more distantly in the intron which cause altered splicing (such as those resulting in cryptic splice site activation and inclusion of pseudoexons into mRNA).
No one technique as yet stands out as definitive for diagnosis in BM and UCMD, but detection of pathogenic changes in one of the COL6 genes remains the gold standard. We have demonstrated that SCAIP is a robust methodology by which this can be approached. An additional advantage of this methodology whereby all genes were screened in all patients is that we have been able to identify potentially novel modes of inheritance and that the full range of polymorphisms in the three genes can be appreciated, which may potentially be of importance for determining the major phenotypic variability seen in this group of disorders, especially as some patients showed changes in more than one of the COL6 genes. The high frequency of polymorphic changes also has implications for further evaluation of primary sequence information. Reliable classification of sequence changes as polymorphisms will reduce the ambiguity of a direct sequence approach as more sequence data accumulate. Details on detected sequence variations are available online at http://www.genome.utah.edu/DMD/collagensnps/.
Acknowledgments
The authors thank the patients and their families for their participation in this study. The authors are grateful to K O’Brien and D Sudano for their technical assistance and thank UC Reed, M van der Knaap, RS Finkel, H Marks, and M Scavina for contributing patients to this study.
REFERENCES
Supplementary materials
Web-only Table
The table is available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [View PDF] - Primer pairs used to amplify and sequence the coding exons of COL6A1-3
Footnotes
-
This work was supported by MDC (Muscular Dystrophy Campaign) financial aid to the Newcastle Muscle Centre, Myocluster and MDC grants to FM as well as NIH grant AR38912 to MLC. KMF is supported by NIH grants M01 RR00064-39 and R01 NS043264-03. CGB is a Pew Scholar in the Biomedical Sciences. He is also supported by an Ethel Brown Foerder award and by the Florence R.C. Murray Fellowship Program at the Children’s Hospital of Philadelphia. AKL gratefully acknowledges funding by a Wellcome Entry Level Fellowship and subsequently by a Patrick Berthoud Fellowship. SL is funded by the MDC.
-
Conflict of interest: none declared.