Article Text

Abstract
Mental retardation is the most frequent cause of serious handicap in children and young adults. The underlying causes of this heterogeneous condition are both acquired and genetically based. A recently performed refinement of the linkage interval in a large Belgian family with mild to severe non-syndromic X linked mental retardation, classified as MRX9, revealed a candidate region of 11.3 Mb between markers DXS228 and DXS1204 on the short arm of the X chromosome. In order to identify the underlying disease gene in the MRX9 family, we established a gene catalogue for the candidate region and performed comprehensive mutation analysis by direct sequencing. A human homologue of the bacterial 23S rRNA methyltransferase Fstj, the FTSJ1 gene, is located within this region and displayed a sequence alteration in the conserved acceptor splice site of intron 3 (IVS3-2A>G) in all tested patients and carrier females of this family. In contrast, it was absent in all unaffected male family members tested. The mutation results in skipping of exon 4 and introduces a premature stop codon in exon 5, probably leading to a severely truncated protein. Our finding indicates that a protein, possibly associated with ribosomal stability, can be linked to X linked mental retardation (XLMR).
- SAM, S-adenosylmethionine
- XLMR, X linked mental retardation
- FTSJ domain
- mental retardation
- methyltransferase
- MRX9
Statistics from Altmetric.com
Mental retardation is the most frequent cause of serious handicap in children and young adults. The underlying causes of this heterogeneous condition are both acquired and genetically based.1 In the case of X linked mental retardation (XLMR), the prevalence has been estimated to be 1 in 500 males. More than 200 different XLMR entities have been described so far.2–4 Of these, 140 have been classified as syndromic forms characterised by specific clinical, biochemical, or neurological features associated with mental retardation. In contrast to this, non-syndromic XLMR families (MRX) are characterised by impairment of cognitive functions with no additional features. A total of 81 MRX families have been reported so far, with partly overlapping linkage intervals.4 Although knowledge about the genetic causes of cognitive impairment has increased during the last few years and the first genes responsible for non-syndromic mental retardation have been described,5 the total number of genes involved in MRX is still a matter of debate.4,6
Recently, our group has refined the linkage interval of a large Belgian MRX family classified as MRX9,7 from Xp21–q13 to Xp11.4–Xp11.22.8 This four-generation family consists of seven affected males, their five non-affected brothers, and five carrier females (fig 1). Patients were diagnosed as affected with mild to severe mental retardation with no additional specific clinical or dysmorphic features. Carrier women were unaffected.

Pedigree of the MRX9 family. Affected males are indicated by black boxes, the obligate carrier females with dotted circles. Affected and unaffected family members tested for the mutation are indicated with an arrow.
METHODS
Subjects
The MRX9 family is Caucasian and resides in Belgium as described.7,8 The pedigree is shown in fig 1. DNA and RNA were extracted from blood samples and immortalised lymphoblastoid cell lines that were established from peripheral lymphocytes using standard protocols.
Mutation analysis
Mutation screening was carried out on genomic DNA and on cDNA. At the genomic level, the screening was performed by exon sequencing with the adjacent intronic sequences using BigDye kits (PerkinElmer, Heidelberg) with separation of the fragments on an ABI capillary sequencer (ABI 3100). For PCR amplification the following primer pairs were used: af (exons 2–4): 5′ -CTT CTG TGT GAG CTG TCA TG- 3′, ar: 5′ -GG CCT TCA CCC AGG ATA TAC- 3′, bf (exon 5): 5′ -CCA GGT AAG AGC ATG GCT G- 3′, br: 5′ -AGG TGA ATA AGT CAA CCA AGG- 3′, cf (exons 6–9): 5′ -ACA AGA AGA TGC ACA GAG CC- 3′, cr: 5′ -ACC TAG ATT GCA TAA GGT GTG- 3′, df (exons 10 and 11): 5′ -CCT GAC CTT GTC CTC AGT TG- 3′, dr: 5′ -CAT AAA GGT TGC CTT ACT CAA AC- 3′. At the cDNA level the coding sequence was amplified with primer pair c1F: 5′ -AGG TGG TAG CCC ATT CAT CTG- 3′ and c1R: 5′ -CTT GTT TGT CCA GGC CTC GTT G- 3′ and sequenced according the above described method. A total of 100 X chromosomes from unrelated healthy males and 200 X chromosomes from 100 unrelated healthy females were tested for the absence of the mutation by DHPLC analysis (WAVE, Transgenomics, Omaha, NE, USA) under the following conditions: gradient of B buffer: 51–61%, running temperature: 65°C. For PCR amplification the following primer pair was used: wa_f: 5′ -CAC GGG CAG TTG ACC TGT G- 3′ and wa_r: 5′ -GGC CTT CAC CCA GGA TAT AC- 3′. Heteroduplex conformation was made possible by adding female wildtype DNA to DNA from hemizygous males.
Northern analysis
cDNA of the FTSJ1 gene was radiolabelled using the Random Primed DNA Labeling Kit (Roche Diagnostics) and hybridised to membranes containing human multiple tissue cDNA panels (MTC panels from Clontech Laboratories, Palo Alto, CA, USA). Hybridisation and washing of the membranes was performed using ExpressHyb hybridisation solution (Clontech) according to the recommendation of the manufacturers.
RESULTS
All known MRX and MRXS genes in the MRX9 linkage interval were excluded as candidate genes (data not shown). In order to identify a novel gene for MRX, a complete gene catalogue was established for the 11.3 Mb large refined linkage interval between markers DXS228 and DXS1204 and mutation analysis of 23 selected candidate genes was performed on genomic DNA and/or at the cDNA level (fig 2A). Genomic sequence analysis of an affected male (IV-1, fig 1) revealed an A>G substitution in the acceptor consensus splice site in intron 3 (IVS3-2A>G) in a human homologue of the bacterial Ftsj gene,9 the FTSJ1 gene (fig 3). This gene is located in Xp11.23, proximal to DXS6949 and distal to DXS6941, between the SN2 and PPN genes (fig 2A). The mutation segregates with the mental retardation in the family, in fact it was found in all affected individuals (III-4, III-12, and IV-1) as well as in obligate carriers II-4 and III-15, while it was absent in the healthy brothers III-9 and III-11 (fig 1). This mutation causes a major transcript significantly shorter than the wildtype fragment (fig 4). Sequencing of the mutant cDNA fragment from the patient revealed skipping of exon 4 that results in a variant predicted to encode for 17 altered amino acids followed by an alternative stop codon at position 335 in exon 5 (fig 3). In addition to the major mutant cDNA fragment that lacks exon 4, an additional faint fragment could be observed in the patient’s sample (fig 4). Indeed, mutations in the acceptor splice can be associated with complex splicing events as demonstrated by our group for an analogous mutation in the BRCA2 gene. There, the substitution of exon 20 -2A>G results in multiple aberrant splice products with clones harbouring deletions of exon 20, clones showing deletions in both exon 20 and 21, and finally clones where exon 20 is deleted together with a part of exon 21 (AM, unpublished data). Cloning experiments are underway to characterise this additional minor variant.

(A) Genes in the refined linkage interval screened for mutations. Genes recently associated with MRX are indicated with a red star. (B) Protein alignment of the FTSJ domain (highlighted in red) in the FTSJ1 protein of various species, showing conservation of the domain. Ce, Caenorhabditis elegans; Dm, Drosophila melanogaster; Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; Sc: Saccharomyces cerevisiae. The abrogated part of the protein caused by the mutation is highlighted in green.

The splice site mutation in FTSJ1. Wildtype FTSJ1 consists of 11 coding exons (yellow), resulting in a predicted 330 amino acids long protein. An intronic A to G exchange in the acceptor-splice site of intron 3 causes skipping of exon 4, resulting in 17 altered amino acids and a subsequent alternative stop codon in exon 5.
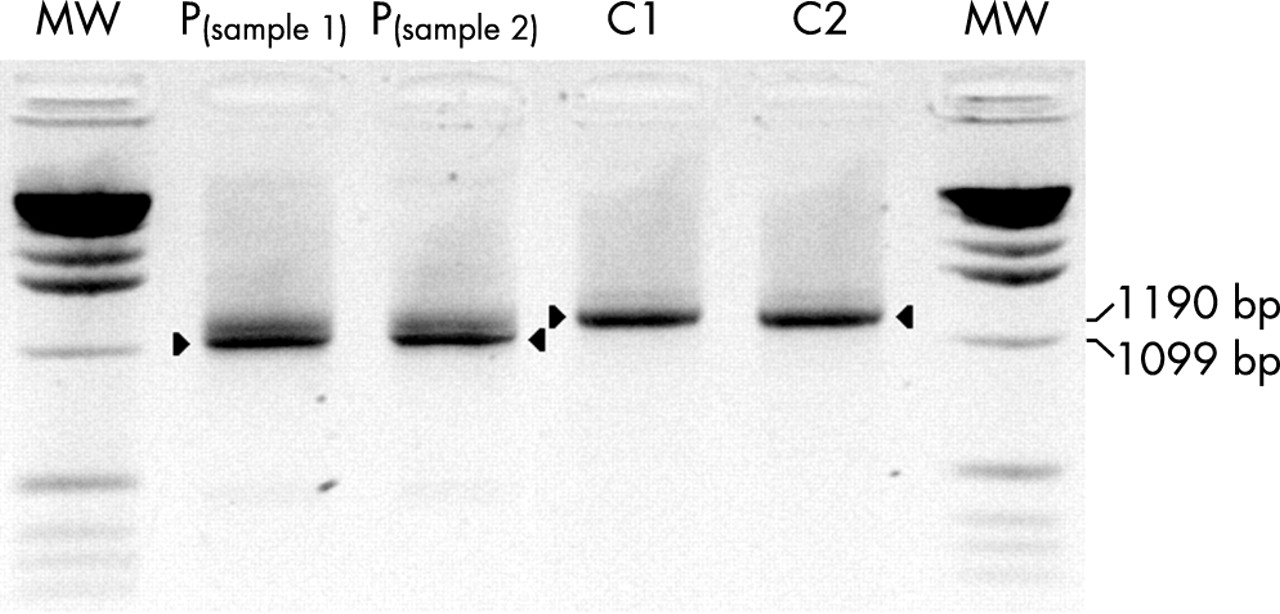
RT amplification of the FTSJ1 gene spanning the 11 coding exons. The amplification of the wildtype transcript results in a 1190 bp product (C1 and C2, lanes 4 and 5), while the transcript of the patient (P, IV-1; samples 1 and 2, lanes 2 and 3) is significantly shorter; it lacks exon 4 as demonstrated by cDNA sequencing (fig 3).
The IVS3-2A>G mutation was not found in 100 unrelated healthy males or in 100 unrelated females (300 X chromosomes). Additionally, we investigated another 32 linked syndromic and non-syndromic XLMR entities and 62 male MR patients, negative for the CGG expansion in the FMR1 gene, for mutations in FTSJ1. However, other than two polymorphisms in introns 6 and 8, no further mutations could be detected in our sample set.
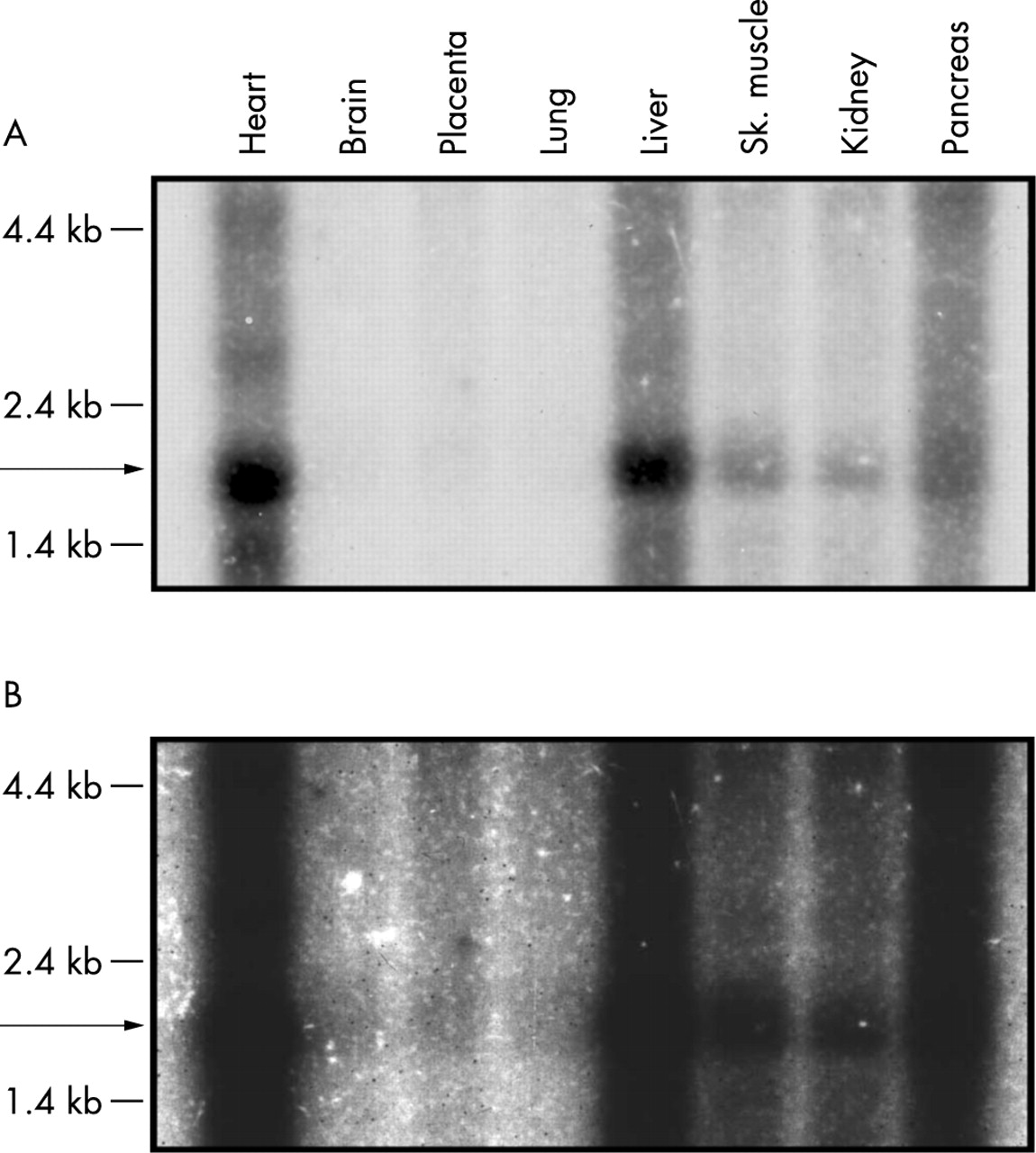
The main transcript of FTSJ1 consists of 13 exons, of which 11 are translated into protein. Mapping and cloning of the whole genomic interval containing this gene was performed by our group recently as part of the German Human Genome Project. The gene was originally submitted by us to the GenBank database as JM23 (accession numbers AF196972 and AJ005892). It is widely expressed but Northern blot analysis revealed highest expression in heart and liver and lower expression in skeletal muscle, kidney, and pancreas. Expression in brain and lung became visible after prolonged exposure (fig 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression pattern of the FTSJ1 gene. A human multiple-tissue Northern blot containing poly(A)+ RNA was hybridised with an RT-PCR fragment of FTSJ1. The 1.9 kb large transcript is indicated by an arrow. (A) Signal intensities after 3 day exposure, (B) signal intensities after 3 week exposure.
DISCUSSION
Our data present the association of a predicted truncated FTSJ1 protein with mental retardation. The predicted wildtype FTSJ1 protein is 330 amino acids long and highly conserved during evolution, with 34% aa identity to the E. coli Ftsj protein.10 In the MRX9 family, a splice site mutation that results in skipping of exon 4 in the FTSJ1 gene, is predicted to cause a truncated protein of 82 amino acids. A highly conserved FtsJ domain of 179 amino acids, common to various bacterial and archaeal transcripts, starts at amino acid position 21 (fig 2B) and is very likely to be severely affected by the predicted truncation. The crystal structure of FtsJ in complex with its cofactor S-adenosylmethionine (SAM or AdoMet) revealed that FtsJ has a methyltransferase fold. The molecular surface of FtsJ exposes a putative nucleic acid binding groove composed of highly conserved, positively charged residues. In E. coli, Fstj is a heat-shock protein that methylates the 23S rRNA at position U2552, using S-adenosylmethionine as a methyl-group donor.11–13 U2552 is a conserved residue in the ribosomal A site, which contacts the aminoacyl-tRNA during protein synthesis.14 Methylation at the ribose 2′-OH group plays an important role in ribosome assembly and functioning. Fstj null mutant bacteria show a dramatically altered ribosome profile and growth retardation.10 In all eukaryotic species examined, three different homologues of Fstj are found, called FTSJ1, FTSJ2 (7p22.3),15 and FTSJ3 (17q23.3) in humans. It is not known whether this family of proteins has the same function in humans as Ftsj in bacteria, but sequence conservation among all members of this protein family is high, especially in the functional domains. Moreover, Spb1P, the yeast homologue of FTSJ1, is required for ribosome synthesis and is able to bind S-adenosylmethionine in vitro, suggesting that it is an active methylase in yeast.16
The physiological and functional roles of the FTSJ1 protein are still obscure, but recent data indicate involvement in either of the following pathways. Overexpression of two small, universally present Rho GTPases, Obg and EngA, restores ribosome assembly and growth disadvantage in Ftsj mutant bacteria, without methylation of the U2552 residue.17 Though the rescue mechanism remains unclear, these data suggest that, at least in bacteria, some interaction between the Ftsj and small GTPase pathways may exist. A second hypothesis regarding the involvement of FTSJ1 in cognitive function is based on its capacity to bind S-adenosylmethionine (SAM). SAM serves as sole methyl donor for methylation processes in the central nervous system and was found to cross the blood–brain barrier. Low cerebrospinal fluid levels of SAM have been observed in several neuropsychiatric and neurological disorders including depression, brain ischaemia, and dementia.18 Careful clinical evaluation of patients from the MRX9 family has not yet revealed symptoms other than mental retardation. This may indicate that the other mammalian FTSJ proteins can compensate for the lack of FTSJ1 in tissues other than the brain. In bacteria, the translational activity is severely impaired and our findings indicate that this process is at least not optimal in mammalian brain. To what extent the other FTSJ proteins are involved in generating ribosomal stability is one of several questions posed by this finding.
Screening of 32 additional syndromic and non-syndromic XLMR entities and 62 males with MR of unknown etiology revealed no additional mutations in FTSJ1. However, another group detected two different truncating mutations in the same gene in two other MRX families (Freude et al, 53rd ASHG meeting 2003, oral presentation). Together with our results these findings strengthen the involvement of FTSJ1 in cognitive impairment. Nevertheless, the low frequency of FTSJ1 mutations in MRX families further supports the heterogenic basis of MRX.
In conclusion, we report the identification of a novel gene associated with MRX. The FTSJ1 gene was mutated in a large four-generation family. To increase our knowledge of the function of FTSJ1 in ribosome metabolism and neuronal processes, additional experiments will include knock-out experiments applying, for example, siRNA in specific neuronal cell systems and the detection of interacting partners by yeast-two hybrid experiments.
Acknowledgments
We would like to thank the members of the MRX9 family.
REFERENCES
Footnotes
-
This work was supported by grants from the German Ministry for Research and Education (BMBF, 01KW9974) and the European community (EC, QLG2-CT-1999-00791) to AM and by grants from the Belgian National Fund for Scientific Research - Flanders (FWO) and an Interuniversity Attraction Poles Program (IUAP-V) to BW and RFK and a grant from NICHD (HD26202) to CES.
-
Conflict of interest: none declared.