Article Text

Abstract
Great advances in the field of genetics have been made in the last few years. However, resolving the complexity that underlies the susceptibility to many polygenic human diseases remains a major challenge to researchers. The fast increase in availability of genetic data and the better understanding of the clinical and pathological heterogeneity of many autoimmune diseases such as multiple sclerosis, but also Parkinson’s disease, Alzheimer’s disease, and many more, have changed our views on their pathogenesis and diagnosis, and begins to influence clinical management. At the same time, more powerful methods that allow the analysis of large numbers of genes and proteins simultaneously open opportunities to examine their complex interactions. Using multiple sclerosis as a prototype, we review here how new methodologies such as gene expression profiling can be exploited to gain insight into complex trait diseases.
- complex traits
- heterogeneity
- multiple sclerosis
- new approaches
Statistics from Altmetric.com
In the view of classical genetics, diseases are divided into Mendelian disorders and complex traits. While the former are attributed to single gene mutations with a simple mode of inheritance, the latter are thought to result from multiple genes, each playing a small and interactive role in the susceptibility to the diseases. From a clinical point of view a continuum of phenotypic presentations may be observed. At one end of the spectrum are disorders caused by fully penetrant deleterious mutations and on the opposite end are environmental diseases. Between these two extremes lie the incompletely penetrant and the polygenic disorders, creating a smooth transition from strictly genetic to multifactorial illnesses (fig 1).1 The identification of the causative genes for Mendelian disorders has been a major undertaking for researchers, in most cases requiring many years of investigations, for example, tuberous sclerosis.2 For obvious reasons, the characterisation of the genetics underlying the complex traits poses substantially greater challenges.3 However, as these are far more common than the Mendelian disorders, tackling the complex traits has substantial socioeconomic impact. Heterogeneity is the common denominator to all of them and can be considered the most important hurdle to overcome. As an example, thinking of cancer as a condition resulting from an alteration of cell cycle control mechanisms would not only oversimplify and unjustly pool together an immense number of aetiologically and pathogenetically distinct disorders, but it would also be unlikely to be successful. Similar phenotypes may be produced by different genes in the same pathways as well as by completely unrelated causes. Tumours may in some cases arise from exposure to environmental agents (for example, asbestosis),4 while in other instances single gene mutations are of primary importance (for example, retinoblastoma).5 By analogy, it appears imperative to identify biomarkers that would allow a stratification of cases into homogeneous classes for the purpose of dissecting the complexity of the multifactorial diseases.6 Subsequently, the analyses performed in the so stratified groups will have much greater chances to show the mechanisms in common to each category. In particular, researchers have applied new techniques to achieve a novel classification of apparently homogeneous groups of tumours, thus permitting new insight into the pathogenesis of some malignancies.7,8 In this review we are referring mainly to multiple sclerosis (MS) as well as to some other autoimmune and neoplastic conditions as models in support of the aforementioned point.

The progressive decrease in the genetic load contributing to the development of a disease creates a smooth transition in the distribution of illnesses on an aetiological diagram. In theory there are no diseases completely free from the influence of both genetic and environmental factors.
THE EARLY STEPS IN MOLECULAR GENETIC INVESTIGATIONS
Before the emergence of the new molecular techniques, epidemiology was the means to evaluate the relative weight of genetic versus environmental factors, and its application to the problem remains very valuable,9 as shown by our example, multiple sclerosis.10 A genetic predisposition to this chronic debilitating neurological illness was initially suspected based on a fairly large number of observations both at the population level and by familial aggregation studies.11 In this context, twin surveys are considered an important source of information.12,13 However, the concordance rate in monozygotic twins should only be viewed as the upper limit of the genetic influence on the disease susceptibility, as underscored by the fact that the concordance rate for tuberculosis, an environmental disease, is also higher in monozygotic than in dizygotic twins.14 In MS, the concordance in monozygotic twins varies between <10% to about 30% across different populations.15–18 This is still far from 100%, a figure only expected yet not always observed in monogenic highly penetrant traits.19,20 Overall, the epidemiological studies in MS suggest that genetics is relevant, but also that the environment exerts a key influence on its phenotypic expression.21 In any event, the numerous surveys performed in MS over the course of the last half a century have prompted major efforts of exploiting the new genotyping techniques to assess linkage in large numbers of MS multiplex families. The eight whole genome screens completed to date10 and a meta-analysis of the data22 have provided the concept that MS represents a multifactorial disorder with high levels of complexity. Moreover, linkage studies have proved to be insufficiently powered to detect the small effects of the many genes involved in polygenic disorders, and, while significantly larger studies could be more informative, they are difficult to perform even in disorders that occur at considerable frequencies.23,24 In the case of MS the prevalence is up to about 100/100 000 subjects. Currently, researchers have moved on to testing parents to address whether the susceptibility allele is preferentially transmitted to their single affected offspring at a higher percentage rate than the 50% expected for a neutral allele, according to the Transmission Disequilibrium Test (TDT).25 Many regions of interest (1p, 6p21, 12p, 17q, 19q3) have been identified in MS using this approach.26 More importantly, however, the large amount of data collected by linkage and TDT underscores the relevance of genetic heterogeneity in MS, identifying it as a key point to be addressed.27
Consistent with this view are the findings by Jacobsen et al.28,29 In a molecular genetic investigation in a German population, these researchers have described a strong association between MS and a point mutation at position 77 of exon 4 of the gene encoding the protein-tyrosine phosphatase, receptor type C (PTPRC), also known as CD45. This polymorphism causes an alternative splicing of the mRNA of this membrane phosphatase, thus altering its isoform expression in immune cells. Heterozygosity for the mutation was initially reported both in linkage and association with the disease in three MS nuclear families.28 More recently the same authors have identified in a fourth German multiplex family another mutation (59 C-A transversion) in exon 4 of the PTPRC gene, responsible for a non-conservative amino acid substitution, which affects maturation, activation, and migration of immune cells.29 The fact that some healthy first degree relatives of the index cases in those families carry the mutation, in addition to the lack of association between the CD45 polymorphism and MS in two case-control studies in North American and Swedish populations,30,31 together point to a basic principle of complex traits: none of the involved genes is per se either necessary or sufficient for the development of the disease.32
It is not surprising then that numerous case-control studies seeking association to candidate genes performed in MS as well as in many other diseases have often yielded contradictory results.33
A further instance with similar conclusions to MS is provided by the search for genetic causes of Parkinson’s disease (PD). The identification of a mutation in the alpha-synuclein gene through linkage analysis in large, multigenerational kindreds34 only accounts for a rare autosomal dominant form of PD, and the genetic heterogeneity creates a situation analogous to Alzheimer’s disease where mutations in the presenilin 1 and 2 genes are only responsible for a small minority of the cases.35 Hence, we believe that new approaches are needed to complement this research. Fig 2 offers a schematic view of the analytical methods mentioned in the paragraph.
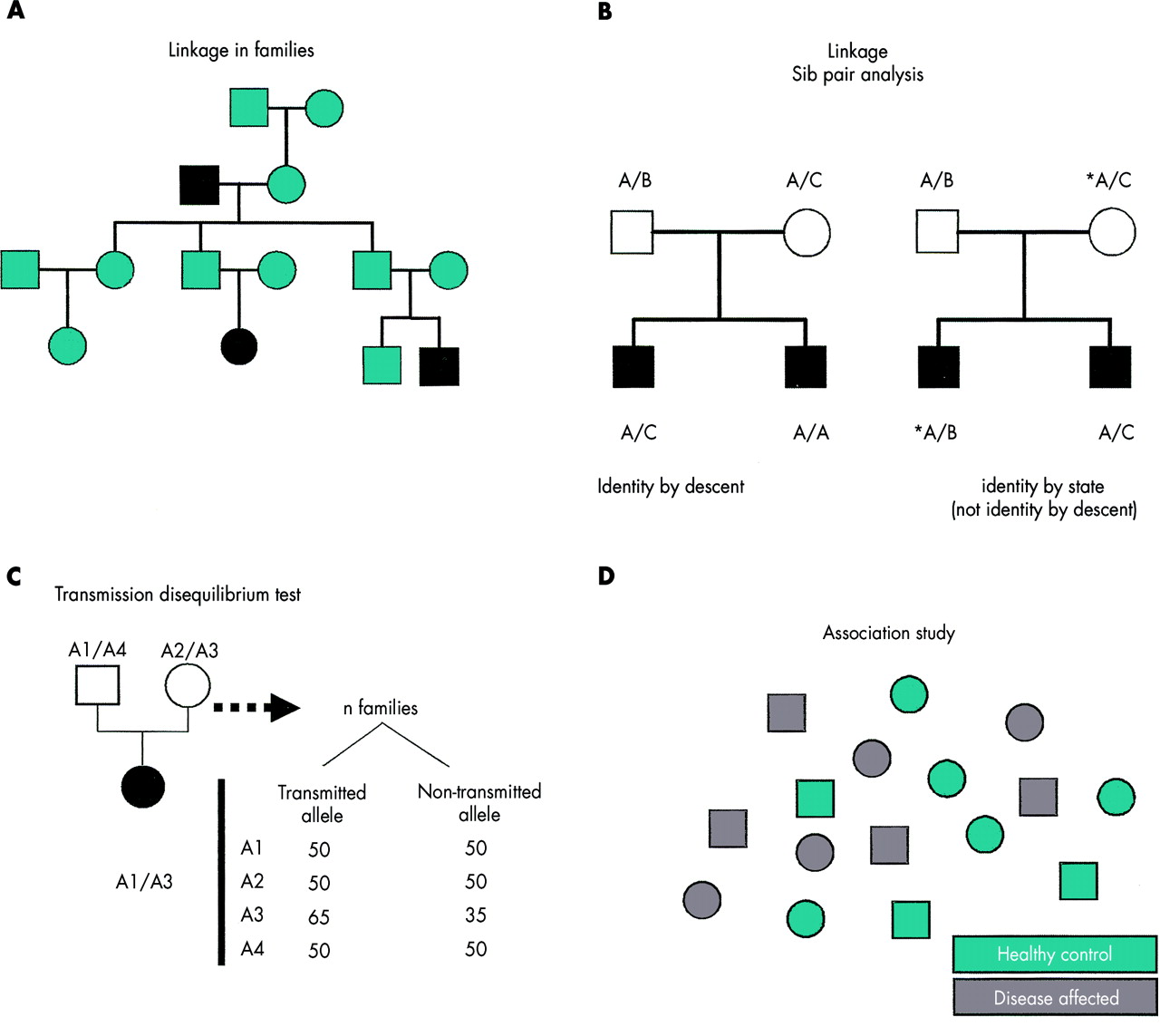
(A) Linkage analysis, aimed at identifying genetic markers that are coinherited with disease status within a multigeneration family, has largely been successful in Mendelian disorders. (B) Sib pair analysis, extensively used in the 90s to investigate the genetics of complex traits, is designed to show alleles that are shared by two or more affected subjects in the same family. (C) The transmission disequilibrium test looks for an allele of a selected marker that is preferentially transmitted to an affected subject by his/her unaffected parents. To be informative the marker has to be polymorphic. (D) Association studies are performed by assessing the statistically significant recurrence of an allele in an affected population in comparison to a control population.
SEEKING “ASSOCIATION” IN AN EXCITING NEW ERA FOR RESEARCH IN GENETICS
Investigators have been puzzled by the question of why two subjects with a similar genetic background would respond so differently, that is, one develops an often serious disorder, while the other remains healthy. In other words, is the major determinant of complex traits a predisposing genetic factor or is an abnormal reaction to external stimuli, acting on epigenetic and stochastic events, more important? This problem is addressed by examining differences between a group of affected subjects and a properly matched healthy control group. These are association type investigations (case-control studies) and are often based on selected hypothesis driven candidate genes.36 However, the interpretation of the results derived from these studies demands caution to avoid false positive and spurious results.37 Yet association studies remain a powerful approach.38 In particular, it is the recent availability of information from the human genome project that has started an era of tremendous opportunities. Initially, it led to the use of multiple tandem repeats (or microsatellites) as markers for disease susceptibility regions, and is now moving forwards to use single nucleotide polymorphisms (SNPs).39 As the SNPs are evenly distributed over the genome and are far more abundant than the microsatellites, efforts are under way to complete the map of the SNPs in the human genome.40 Moreover, haplotypes of SNPs in humans may represent a substantial shortcut in the search for susceptibility genes over the single marker based approaches (fig 3).41 But what is the rationale behind the use of the SNPs in the search for the susceptibility to diseases? The idea is that changes of single base pairs in genomic DNA occur at a predictable rate, that is, the younger a population the fewer the polymorphisms. However, some hot spots in the genome appear to be remarkably more inclined to bear polymorphisms than other more stable areas that are well preserved even throughout many species.42,43 While a deleterious mutation would abrogate a gene function, other more “benign” changes in the coding as well as in the non-coding part of a gene may account for a variation in the functional level of the gene product. It is conceivable that the genetic polymorphisms and their multiple interactions are the basis for the quantitative characters44; they underlie the biodiversity and are therefore attributable to the success of adaptation. Nevertheless, they represent those common variations in the human genome that may affect the predisposition to a disease, and it is worth mentioning that what may be an advantageous profile in one environment, may become unfavourable or uncontrolled in a different setting. Hence, the goal of an association study is to identify those polymorphisms that only exist in an affected population.
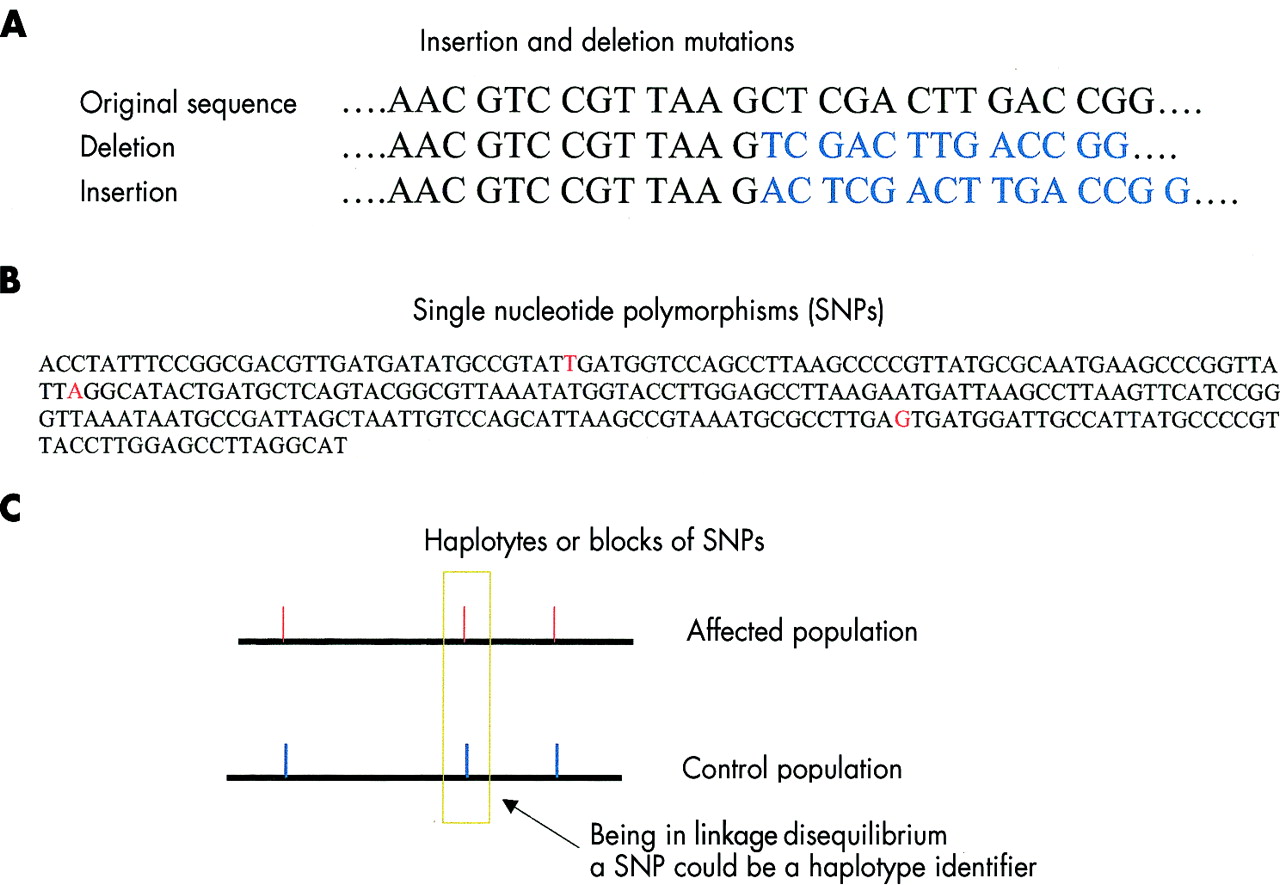
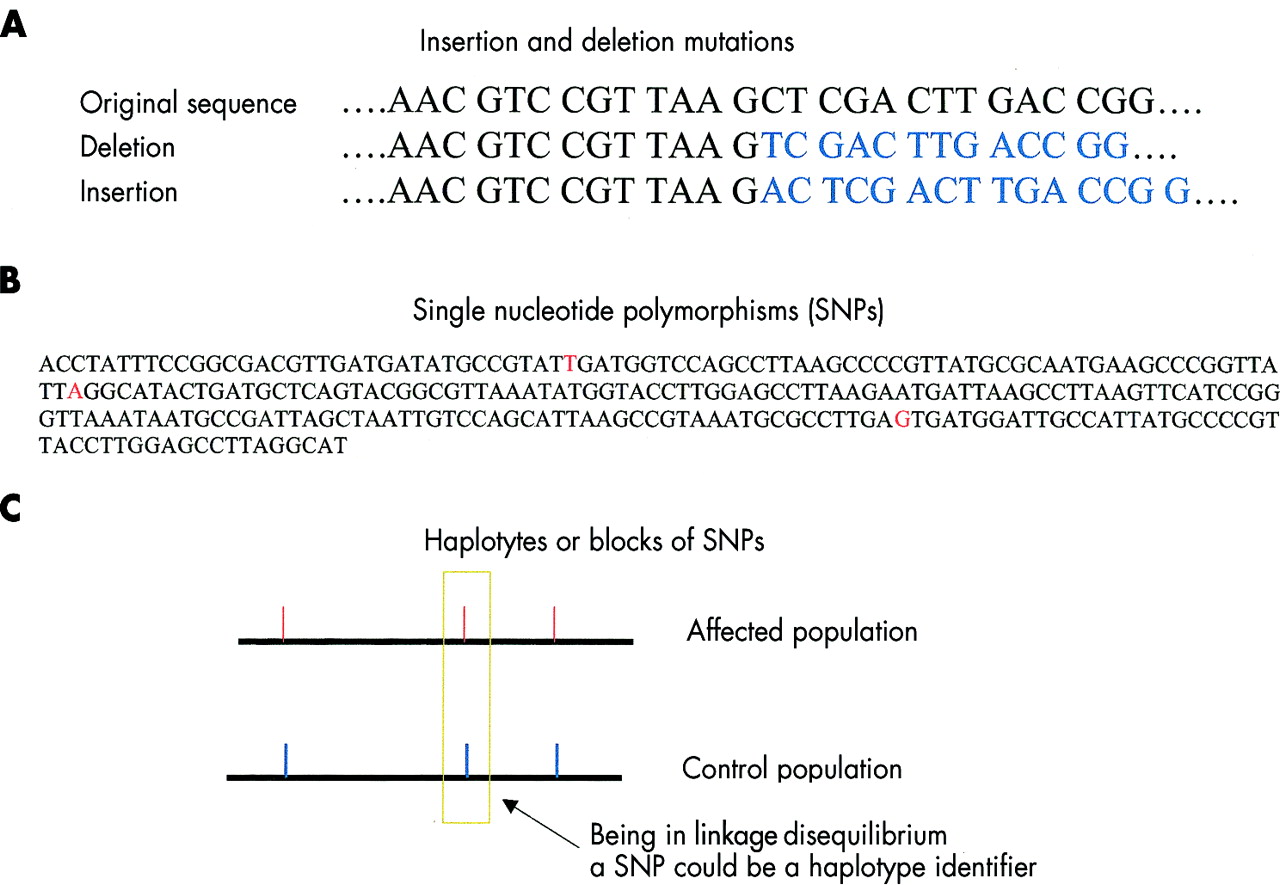
As opposed to base pair insertions and deletions that cause frameshift mutations in a DNA stretch (A), SNPs are single base pair changes that do not necessarily alter the amino acid sequence in the relative gene product, that is, the protein. SNPs (B) recur roughly once in every 1300 bp, and are considered useful markers for genotyping purposes. Haplotypes or blocks of SNPs may be identified in genomic regions where recombination occurs at a low rate and linkage disequilibrium is conserved (C).
Particularly emblematic of complex traits is Hirschsprung disease, or aganglionic megacolon. Several loci have been implicated in the susceptibility to this disease, characterised by the congenital absence of ganglia in various portions of the intestine. Interestingly, distinct genes with diverse mode of inheritance (from dominant to recessive) as well as a full spectrum of degrees of penetrance have been identified by linkage in families and subsequent mapping. Epistatic interactions between genes have also been established to take place, creating a complicated picture of a multiform oligogenic trait that paradigmatically highlights the above notions.45
In regard to the search for polymorphisms responsible for the susceptibility to adverse conditions, one of the most sophisticated examples is given by the immune system whose function is under the control of many interactive genes and environmental factors. In fact the human immune system has evolved over a long period of time, constantly challenged and shaped by external agents. Sudden and dramatic changes in the environment have raised problems of adaptation, as indicated by the steep increase of allergies and autoimmune disorders observed in industrialised countries during the last few decades. According to the hygiene hypothesis, an imbalance in the cytokine production by different T lymphocyte subclasses accounts for the higher incidence of allergies.46 Or, as a more recent theory proposes, increases in both allergy and autoimmunity are attributed to a dysfunction of innate immunity.47–50 While the true causes are not clear at present, sudden changes (certainly remarkable on the evolutionary scale) in daily challenges that the immune system faces in modern societies are possibly responsible for these pathologies, thus the hypothetical point of a lack of adaptation remains valid.
Heterogeneity is a major obstacle to the resolution of the aetiologies of complex traits. One of the best arguments in favour of MS being an autoimmune disease is its linkage to the major histocompatibility complex (MHC, HLA in humans) class II region and association with HLA-DR and -DQ alleles.51–53 It has been estimated that about 10% of the overall genetic susceptibility to MS is conferred by a gene or genes in this area, thus leaving a substantial fraction of susceptibility to be explained by other genes and factors.54 The abovementioned investigations have shown that MS is indeed a polygenic trait. To address the issue of the interaction between genes, Coraddu et al55 used data from a genome wide scan, in this instance stratifying the patients according to HLA status, and repeating the linkage analysis.56 This step resulted in significant changes in linkage scores. Some chromosomal regions that had appeared earlier were not evident any more, whereas new ones emerged. They speculate that these alterations correspond to genes that interact with the MHC region in conferring genetic susceptibility to MS. Furthermore, they conducted an association study in which they observed a correlation between female gender and age of onset and HLA-DR2, but no other features.57 Many other groups have also looked for a correlation between disease phenotype and surrogate markers with partial success.58–60 In fact, the relationship between genotype and phenotype is not always immediately evident, owing to the existence of both genetic (different genes-identical phenotypes) and disease heterogeneity (identical genes-different phenotypes).61 The example of the autoimmune lymphoproliferative syndrome (ALPS) serves to illustrate the case. This disease is characterised by the massive proliferation of germinal centre lymphocytes owing to a defect in the pro-apoptotic signalling molecule FAS (ALPS1a) or its ligand (ALPS1b).62,63 In addition to and as one consequence of the excessive lymphocyte proliferation, the syndrome has autoimmune characteristics, such as manifestations of haemolytic anaemia and thrombocytopenia.64 Genetic heterogeneity is observed when considering ALPS2, a syndrome caused by mutations in the Caspase10 gene, whose product acts along the same pathway downstream of FAS.65 Thus, ALPS provides the case for a multiform disorder in which similar clinical features are determined by single and distinct gene mutations. In addition, even though ALPS may not be considered a classical autoimmune disease, it offers important insights into the pathogenetic mechanisms underlying the breakdown of tolerance.66
The possibility that excessive lymphocyte proliferation leads to autoimmunity has recently been further explored in animal models of systemic lupus erythematosus (SLE). By knocking out either the Gadd45a, a p53 effector gene, or the p21 cell cycle inhibitor, researchers have generated mice that show features typical of SLE.67,68 Similarly to ALPS, the phenotypes in these animals involve deregulated cell proliferation as a result of the loss of functions of Gadd45 and p21 proteins. The possible explanation for the autoimmune component observed in these models is an increased number of autoreactive lymphocytes. Small numbers of autoreactive cells are part of the healthy immune system,69,70 but an excess can overcome the peripheral control mechanisms, rendering the occurrence of the breakdown of tolerance much more probable. Interestingly, female mice defective for either Gadd45a or p21 are significantly more prone to an aggressive and early onset of disease than their male counterparts, a fact that is consistent with the observation of the predominance of many human autoimmune disorders in the female gender, for example, MS. It also underscores that a single gene mutation requires interaction with other factors (unique to females for instance) for the disease to manifest or at least to contribute to its development, and more data of distinct patterns between genders are starting to be generated.71
In light of the above studies, it is clear that cell cycle progression regulators need to be taken into consideration in the context of autoimmune diseases, just as genes controlling lymphocyte activation, cell-cell interaction, or compartment trafficking have been in the past.72 Recent findings by Maas et al73 in various groups of autoimmune disorders further support this prospect. Accordingly, the involvement of the immune system in a multifactorial trait may complicate the search for individual genes predisposing to that disease, as the regulation of the immune system itself is complex and incompletely understood.
NEW TECHNOLOGIES PROVIDE NOVEL INSIGHTS
Understanding the specific events and their sequences occurring during a pathological process is important for many reasons, the ultimate being the identification of an aetiological rather than a symptomatic cure. It is our incomplete knowledge of the disease mechanisms that sets the limits to tailoring specific and effective treatments.74 Yet we believe that the most powerful approach to identifying the causes of diseases is to study well defined categories of patients or subgroups within a disease spectrum. Above, we reviewed how a number of distinct pathogenetic mechanisms may determine apparently analogous conditions, and that defining groups of patients only on the basis of phenotypic characteristics may not suffice. Now, advances in biotechnology provide new tools to researchers at an extraordinary rate,75–80 and it is through the combination of new techniques and alternative approaches that optimal results can be achieved (fig 4). It has been shown that gene expression profiling via microarray experiments is capable of reclassifying apparently homogeneous groups of clinical entities.81,82 Indeed, by interrogating the coordinated expression of large number of genes, this method has already provided new insights into disease mechanisms and led to envisioning strong clinical applications.83,84 Furthermore, by linking gene expression data to existing databases containing functional annotations (such as GeneOntology at http://www.godatabase.org/dev/database/), it is possible to evaluate results from microarray experiments from a functional point of view. Identifying patient groups that are homogeneous at the molecular level, that is, by gene expression profiling, will be facilitated by the creation of large databases containing comprehensive expression data from a variety of tissues and pathological conditions. The importance of this latter goal has convinced and moved investigators and institutions to undertake major efforts to carry out the project and to make the databases immediately available to the public domain.85–88
{kind=link}
{kind=link}
{kind=link}
{kind=link}
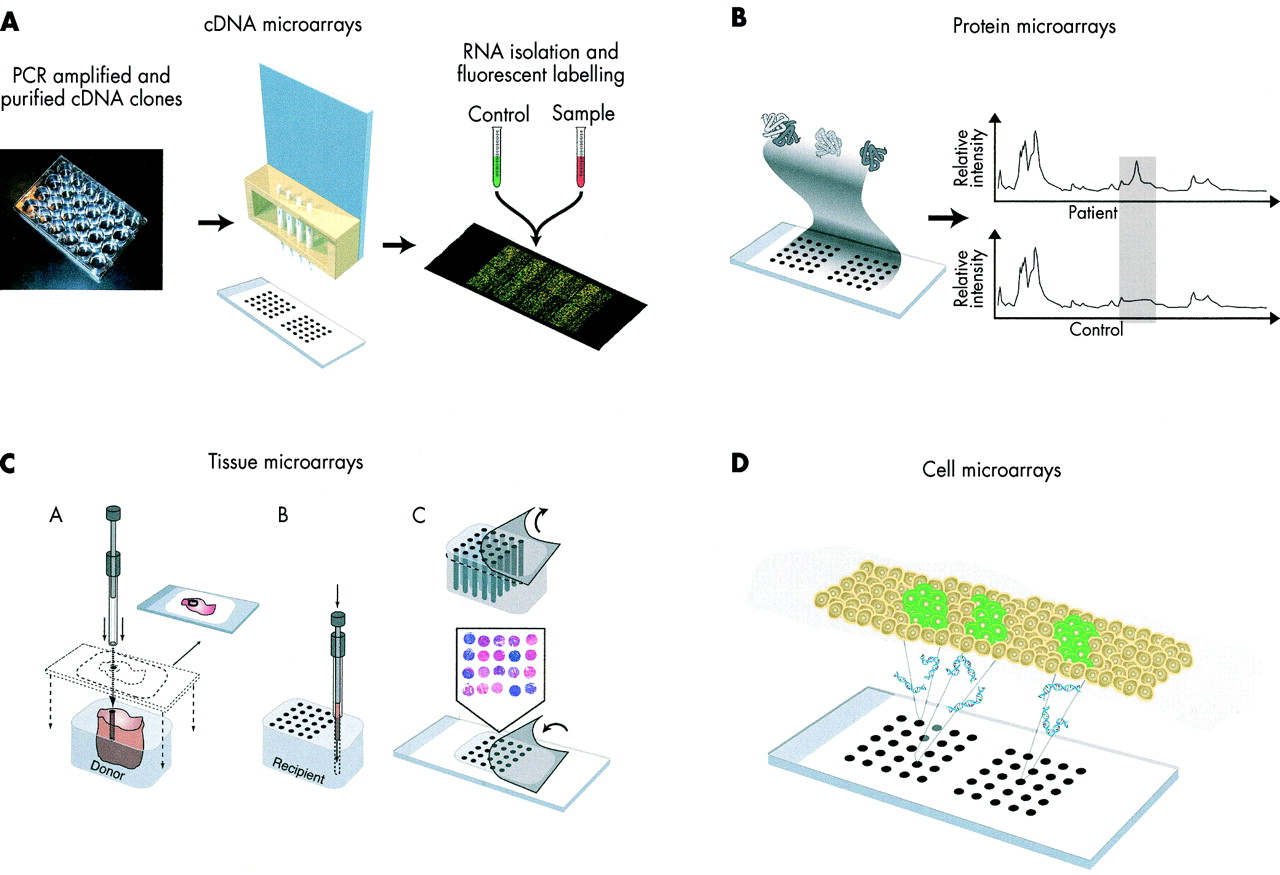
(A) Expression microarray techniques exploit complementary hybridisation, which is a basic property of the nucleic acids. Large numbers of DNA probes are deposited in high density on glass slides or membranes. These probes are either robotically spotted PCR amplified products of cloned cDNAs or synthesised oligonucleotides, or alternatively on glass synthesised oligonucleotides (such as Affymetrix Genechips). The RNA sample to be tested (or the genomic DNA in the case of the CGH) can be labelled with various methods and is assayed onto the microarray platform. Only the spots containing the specific sequence present in the samples will “light up” proportionally to the amount in the original sample. (B) Protein arrays are designed to detect the presence in a given biological fluid of proteins that may interact with those deposited on the array, probes. Probes can be antigens, antibodies, other binding proteins, or DNA with a specific protein binding motif. In most cases the identity of the positive spots, whose readout is expressed in terms of peaks, remains unknown. (C) In the tissue microarray, small tissue samples are punched from a pathology specimen embedded in a donor paraffin block (A) and arrayed into a recipient block together with a high number of other tissues to be examined in parallel (B). Subsequently, the recipient block is horizontally sliced and each slice is transferred onto the test slides (C) to yield a histological slide comprising up to 1000 samples in each. (D) The most recent evolution of the array based technology is the cell array. Here molecules such as antisense oligonucleotides or small interfering RNA (siRNA) are deposited onto glass slides. Living cells are then grown on the slides, resulting in gene silencing via reverse transfection that occurs in the area spotted with the gene specific target sequence. Functional assays such as reporter assays involving green fluorescent protein are used to identify the biologically relevant gene.
The availability of microarray technology has led our group to study MS genetics by profiling gene expression in peripheral blood mononuclear cells of patients and healthy subjects in order to assess whether it allows the distinction of patients from controls (unpublished data). The rationale behind the use of peripheral blood cells is in the possibility that primed and activated effector cells relevant to the pathogenesis of MS may circulate in the periphery, resulting in a better understanding of the pathogenesis of the disease, as recently suggested.89 In line with previous studies by Ramanathan et al90 and others,73 we found that subtle differences in the level of expression of various immune and cell cycle related genes are detectable in the MS patients when compared to controls. However, as expected for a complex trait, we noticed that none of the genes works as a perfect discriminator of the two groups. Assuming that the gene expression reflects the genetic predisposition to the disease rather than being the altered response to an external agent, it is conceivable to explain our finding on the basis of the existence of genetic heterogeneity in the MS population. This would be in accord with observations in recent years that MS is a heterogeneous group of disorders in many aspects, including clinical presentation, course and severity, histopathological characteristics of lesions, magnetic resonance imaging findings, response to treatment, and immunological observations.51,91,92 It has been shown that measuring multiple immunological parameters, stratifying patients according to MRI characteristics, and incorporating clinical parameters allows the recognition of subgroups among MS patients.93,94 This approach could be further improved by the combined use of microarrays, MRI scans, and in the near future proteomics data.
CONCLUSIONS
The classification of complex traits into well defined categories is the prerequisite for understanding the underlying pathogenetic mechanisms. This can only be achieved by using the most sophisticated tools available applied to large numbers of samples. Techniques that simultaneously analyse as many genes or proteins as possible have already been put into practice, and as the collection of appropriate sets of samples is an essential step in this endeavour, a close interaction between the medical and the research community is crucial. To accomplish a successful search and to address the complexity of the multifactorial disorders a joint effort is required, particularly to assure a collection of reliable data before they can be used for patient management.95 Like assessing the degree of polymorphism in a given population, which involves genotyping individual subjects,96 we have to analyse distinct patient cohorts if we aim at characterising subsets of MS patients to define the disease phenotypes. Or, maybe, the more we comprehend about disease pathogeneses, the clearer it becomes that the need for classification is only ours not Mother Nature’s.
Acknowledgments
We would like to thank M Cichanowski of the Scientific Illustration Unit, NIH, for his invaluable help with the illustrations.