Article Text

Abstract
Five missense mutations in the FCH/LCCL domain of the COCH gene, encoding the protein cochlin, are pathogenic for the autosomal dominant hearing loss and vestibular dysfunction disorder, DFNA9. To date, the function of cochlin and the mechanism of pathogenesis of the mutations are unknown. We have used the biological system of transient transfections of the entire protein coding region of COCH into several mammalian cell lines, to investigate various functional properties of cochlin. By western blot analysis of lysates prepared from transfected cells, we show that cochlin is a secreted protein. Immunocytochemistry shows concentrated localisation of cochlin in perinuclear structures consistent with the Golgi apparatus and endoplasmic reticulum, showing intracellular passage through these secretory compartments. We detected that cochlin is proteolytically cleaved between the FCH/LCCL domain and the downstream vWFA domains, resulting in a smaller cochlin isoform of ~50 kDa. Interestingly, this isoform lacks the entire mutation bearing FCH/LCCL domain. We have also shown that cochlin is N-glycosylated in its mature secreted form. Previous studies of the FCH/LCCL domain alone, expressed in bacteria, have demonstrated that three of four DFNA9 mutations cause misfolding of this domain. Characteristic eosinophilic deposits in DFNA9 affected inner ear structures could be the result of aberrant folding, secretion, or solubility of mutated cochlins, as in certain other pathological states in which misfolded proteins accumulate and aggregate causing toxicity. To examine the biological consequences of cochlin misfolding, we made separate constructs with three of the DFNA9 mutations and performed parallel studies of the mutated and wild type cochlins. We detected that mutated cochlins are not retained intracellularly, and are able to be secreted adequately by the cells, through the Golgi/ER secretory pathway, and also undergo proteolytic cleavage and glycosylation. These results suggest that DFNA9 mutations may manifest deleterious effects beyond the point of secretion, in the unique environment of the extracellular matrix of the inner ear by disrupting cochlin function or interfering with protein-protein interactions involving the FCH/LCCL domain. It is also possible that the mutations may result in aggregation of cochlin in vivo over a longer time course, as supported by the late onset and progressive nature of this disorder.
- COCH
- cochlin
- DFNA9
- deafness and vestibular disorder
Statistics from Altmetric.com
In the last decade, enormous progress in the discovery of genes involved in hearing and deafness has been made through identification of many new loci and specific mutations causing heritable deafness and vestibular disorders.1 Functional studies to elucidate the roles of these genes in the process of hearing and balance maintenance and the molecular events underlying the pathology resulting from mutations in these genes are continuing. One such gene, COCH (coagulation factor C homology),2,3 encodes the protein cochlin, which is mutated in the autosomal dominant deafness and vestibular disorder designated DFNA9.4–7 Cochlin mutations have also been implicated in certain cases of Meniére disease (with features of hearing loss and vertigo)6 and may possibly be associated with presbyacusis (age related hearing loss),5 common disorders that are probably heterogeneous and multifactorial in aetiology. Whether COCH mutations are validated to be aetiological or common in either Meniére disease or presbyacusis warrants further investigation in a larger series of patients.
Mature cochlin (fig 1) is a modular polypeptide consisting of an N-terminal factor C homology domain (FCH), and two von Willebrand factor A-like domains (vWFA1 and vWFA2). All identified DFNA9 missense mutations fall in the same region, the FCH domain of cochlin. These mutations were found in families from the United States, Australia, Belgium, and The Netherlands with DFNA9 deafness and vestibular disorder (table 1).4–7 The FCH domain (also referred to as the LCCL domain for its presence in Limulus factor C, cochlin, and late gestation lung protein) is a novel fold consisting of a central α helix wrapped by two β sheets composed of a total of eight β strands.8,9 The function of the FCH/LCCL in these various proteins is still unknown; adhesive properties and roles in the extracellular matrix, coagulation, or host defence mechanisms have been proposed.8,10
COCH mutations in DFNA9 families

Schematic representation of the deduced amino acid sequence of human COCH, encoding the protein cochlin, shows a predicted signal peptide (SP), followed by a region homologous to a domain in factor C of Limulus (FCH, factor C homologous domain), an intervening domain (ivd1), and two von Willebrand factor A-like domains (vWFA1 and vWFA2) separated by a second intervening domain (ivd2). Five known missense mutations in the FCH domain, causing the DFNA9 deafness and vestibular disorder, are indicated by black arrows below the schematic drawing. The positions of all cysteine residues are shown. Mutant variants generated for transient transfections are indicated with an asterisk. Positions of the N-terminal FLAG tag and C-terminal HA tag are indicated by white arrows above the schematic drawing. Most experiments were performed with constructs which only have the HA tag. The addition of the FLAG tag to the HA tagged constructs was done subsequently, to detect the N-terminal polypeptide product following proteolytic cleavage.
Most cochlin mutations causing DFNA9 are located on the surface of the FCH domain, but when the FCH domain is expressed in E coli, their effect is to interfere with proper folding of this domain.9 Specifically, three of the four point mutations tested (P51S, V66G, G88E) resulted in misfolding and aggregation of bacterially expressed FCH domains.9 Interestingly, a very characteristic histopathological finding in DFNA9 is the accumulation of large amounts of eosinophilic acellular material in the cochlear and vestibular labyrinths,11–13 in areas where cochlin mRNA and protein are expressed.14 Electron microscopic examination of these deposits shows dense, highly branched and disarrayed, non-parallel microfibrils with a granular substance (possibly glycosaminoglycans) scattered among the fibrils.12 These findings raise the possibility that misfolding of mutated cochlins in affected patients might lead to intracellular and/or extracellular cochlin deposition.
Mutations causing protein misfolding may manifest with aberrations of protein processing, stability, or secretion in cultured cells, which provide a convenient system to study pathophysiological mechanisms. We have used transient transfections of COCH constructs in mammalian cell lines as a biological system to study various functional parameters of cochlin, such as subcellular localisation, secretion, glycosylation, and post-translational processing. We have performed parallel studies of the normal and three mutant forms of cochlin (P51S, V66G, G88E) to investigate these properties and to assess any differences in subcellular trafficking, secretion, glycosylation, proteolytic processing, and protein stability.
MATERIALS AND METHODS
Normal and mutated cochlin expression constructs
Human wild type COCH cDNA (GenBank Accession No AF006740) containing its entire protein coding region (550 amino acid residues) and ~30 bp of 5′ untranslated region was cloned into a modified form of pcDNA3 (Invitrogen, Carlsbad, CA) that permits expression of polypeptides with three C-terminal haemagglutinin (HA) tags. PCR primers with appropriately introduced restriction sites were used to amplify and clone the COCH cDNA in frame with C-terminal HA tags. The COCH cDNA was also cloned into the same vector with its stop codon, to generate untagged full length coding COCH. Constructs were sequenced to exclude PCR errors.
DFNA9 constructs were generated by introducing point mutations into the normal COCH sequence, using QuikChange site directed mutagenesis following the manufacturer’s protocol (Stratagene, La Jolla, CA). Three (out of five known) missense mutations (table 1) causing DFNA9 were generated: CCA to TCA (Pro to Ser at codon 51, designated P51S), GTA to GGA (Val to Gly at codon 66, designated V66G), and GGA to GAA (Gly to Glu at codon 88, designated G88E). An HA tagged construct from an unrelated gene, intracellular domain of NOTCH (ICN), was used as a positive control in transfections, Western blot analysis, and immunocytochemistry.
A FLAG epitope tag was introduced subsequently into the normal and mutated COCH constructs (which already have the HA epitope tag) between the signal peptide and the beginning of the FCH domain, by insertion of the tag, using QuikChange site directed mutagenesis following the manufacturer’s recommendations (Stratagene).
Cell culture and transient transfections
293T (human embryonic kidney), COS7 (African green monkey kidney), and NIH3T3 (mouse fibroblast) cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units of penicillin, and 100 μg of streptomycin in 60 mm dishes (harvested for western blot analysis) or Permanox Lab-Tek chamber slides (Nalge Nunc, Naperville, IL) (for immunocytochemistry). Cells were transiently transfected in the absence of antibiotics with equal quantities of normal and mutated COCH plasmids using Lipofectamine 2000 transfection reagent (for 293T and COS7 cells) or Lipofectamine Plus reagent (for NIH3T3 cells) according to the manufacturer’s protocols (Invitrogen).
Immunocytochemistry
Cells on chamber slides were fixed in 3% paraformaldehyde in PBS for 15 minutes and then washed five times briefly in PBS. Blocking of non-specific sites was done with 4% non-immune goat serum in PBS for five minutes, followed by incubation for one hour at room temperature with the primary antibody, mouse anti-HA monoclonal antibody (clone HA.11) (Covance, Richmond, CA) in PBS containing 1% goat serum and 0.1% NP40. In some experiments, NP40 was omitted. Cells were washed five times in PBS, incubated for five minutes in blocking solution, and then incubated for one hour at room temperature with the secondary antibody, fluorescein (FITC) conjugated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA). After washing five times in PBS, slides were mounted with Vectashield mounting medium with DAPI to stain nuclei (Vector Laboratories, Burlingame, CA). Before mounting, some slides were incubated with tetramethylrhodamine B isothiocyanate (TRITC) conjugated phalloidin (Sigma, St Louis, MO) for binding to filamentous actin, and washed five times in PBS before applying coverslips. Slides were visualised and images captured using an Olympus AX-70 fluorescence microscope and Photosys CCD camera (Applied Imaging, Santa Clara, CA). Composite images were generated using Genus software (Applied Imaging).
Immunoprecipitation
Conditioned media (300 μl to 1 ml) were incubated with anti-HA (clone HA.ll) bound to beads (Covance) for two hours at 4°C. Beads were washed three to five times in PBS, followed by solubilisation of adsorbed proteins with SDS-PAGE loading buffer with or without β-mercaptoethanol (β-ME) at 95–100°C for five minutes. Alternatively, for subsequent digestion with glycosidases (New England Biolabs, Beverly, MA), proteins were solubilised in the manufacturer’s glycoprotein denaturation buffer (0.5% SDS, 1% β-ME) at 95–100°C for five minutes. Immunoprecipitations were also performed on media of cells transfected with FLAG tagged COCH, using anti-FLAG M2 affinity gel (purified murine IgG monoclonal antibody covalently attached to agarose) (Sigma).
Glycosidase digestion
HA tagged secreted wild type and mutant cochlin proteins, immunoprecipitated from the media of transfected cells, were digested with N-glycosidases, PNGase F (N-glycosidase F) and endoglycosidase H according to the manufacturer’s protocol (New England Biolabs).
Western blot analysis
After transfection of cells with wild type and mutant COCH constructs, the medium was centrifuged to remove any detached cells, and secreted proteins were immunoprecipitated as described above. Cells were washed with cold PBS, centrifuged, and lysed in SDS-PAGE loading buffer. Cell lysates, as well as immunoprecipitated proteins from media, were heated at 95–100°C for five minutes in SDS-PAGE buffer, separated by 10% SDS-polyacrylamide gel electrophoresis, and transferred to Immobilon-P (Millipore, Bedford, MA) membranes using either a wet transfer apparatus (Idea Scientific, Minneapolis, MN) or the Trans-Blot SD semi-dry transfer apparatus (Bio-Rad, Hercules, CA). For detection of the small N-terminal cochlin peptide (~10 kDa estimated size) predicted to be the product of post-translational proteolytic cleavage, proteins were separated on 15% polyacrylamide gels and transferred onto 0.2 μm pore size membranes. Membranes were incubated in blocking buffer (5% non-fat dry milk, 0.075% Tween 20 in PBS) for one hour at room temperature, washed four times for 10 minutes in PBS and 0.075% Tween (PBS-T), and incubated for one to two hours with the primary antibody, mouse anti-HA monoclonal antibody (clone HA.11) (Covance). Alternatively, blots were incubated with a polyclonal rabbit anti-human cochlin (anti-FCH/cochlin) raised against a small peptide (amino acid residues 34–48) in the FCH domain of cochlin (gift of Maria Kamarinos and Henrik Dahl, The Murdoch Children’s Research Institute, Royal Children’s Hospital, Melbourne, Australia).
Membranes were washed four times for 10 minutes in PBS-T, and then incubated with peroxidase conjugated goat anti-mouse (DAKO Corporation, Carpinteria, CA) or goat anti-rabbit immunoglobulins (DAKO Corporation) for one hour. After washing four times for 10 minutes in PBS-T and incubation for one to five minutes in Super Signal Chemiluminescent Substrate Stable Peroxidase and Luminol Enhancer (Pierce, Rockford, IL), autoluminograms were prepared.
RESULTS AND DISCUSSION
Subcellular localisation of normal and mutated cochlins
The predicted amino acid sequence of COCH has an N-terminal putative signal peptide and no other apparent hydrophobic regions consistent with transmembrane domains, indicating that cochlin is likely to be a secreted protein. To assess intracellular localisation and secretion of cochlin, we performed immunostaining of mammalian cells transfected with wild type and mutant COCH constructs (fig 2). Prominent staining for cochlin is seen in the endoplasmic reticulum (ER)/Golgi network adjacent to the nucleus. More diffuse staining is also detected throughout the cytoplasm. These results indicate that cochlin goes through the ER/Golgi secretory pathway, a finding that is corroborated by detection of cochlin in the culture media of transfected cells. Furthermore, we show that cochlin is glycosylated in its mature secreted form, also supporting its transport through the Golgi apparatus.

Immunocytochemistry showing subcellular localisation of cochlin after transient transfections of (A) NIH3T3 and (B, C) COS7 cells, with wild type HA tagged human COCH. Cochlin was detected with primary anti-HA antibody and FITC conjugated secondary antibody. (A) Cochlin localisation is seen as green immunofluorescence in perinuclear structures consistent with the Golgi apparatus and endoplasmic reticulum in (A) NIH3T3 and (C) COS7 cells; diffuse staining is also detected throughout the cytoplasm. Nuclei are stained in blue with DAPI. No immunostaining for cochlin was detected in the nucleus. (B) The actin cytoskeleton is visualised in red, by staining with rhodamine conjugated phalloidin, which binds filamentous actin. (D-F) Transient transfections of COS7 cells with HA tagged mutant COCH constructs show a pattern of localisation similar to the wild type. The following missense mutations in COCH, which cause autosomal dominant sensorineural deafness and vestibular dysfunction, were tested: (D) V66G, (E) G88E, (F) P51S.
Immunostaining of the three mutant forms of cochlin in transfected cells shows the same perinuclear localisation as seen for wild type, indicating that they are transported also through these structures. They do not appear to be subject to accumulation or degradation as seen in other precedents of disease states in which mutant proteins may exhibit different localisation or cellular trafficking, or may undergo degradation or aggregation as a result of misfolding. Varying degrees of protein misfolding occur “normally” in cells during the course of protein synthesis, owing to errors in synthesis or assembly.15 The cell has several “quality control” mechanisms to correct or eliminate misfolded proteins and intermediates, including chaperone mediated refolding and proteosomal degradation.15 In the case of severely misfolded proteins, these control mechanisms may lead to retention and degradation. For example, the common ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) results in retention in the ER, extensive degradation, and a lack of any functional protein.16 Misfolded proteins may also accumulate in “holding stations” called aggresomes,15 dynamic structures that may take the form of cytoplasmic inclusions. Protein aggregates or aggresomes are a prominent feature in a number of pathological states, including Alzheimer’s disease (β-amyloid precursor protein),17 Huntington’s disease (huntingtin),18–20 Parkinson’s disease (α-synuclein),21 and amyotrophic lateral sclerosis (superoxide dismutase).22,23
Previous results showing misfolding as a result of COCH mutations9 were done using only the FCH/LCCL domain and not the full length protein including the other cochlin domains; furthermore, the in vitro misfolding of this domain was assessed by its expression in bacteria. Our studies, however, are performed in the biological system of cultured mammalian cells, using the entire coding sequence of cochlin for transfection, therefore reflecting possible effects of the other domains of cochlin on the mutation bearing domain, as well as the influence of cellular mechanisms which would represent in vivo processes more accurately. The absence of overt intracellular retention or degradation of mutated cochlins in the transfected cells suggests that if there is indeed misfolding of the FCH domain within the cellular system and in the context of the entire protein, it may be less prevalent in cultured mammalian cells than as previously shown in bacteria, or may not be sensed by quality control mechanisms of the cell.
For localisation of wild type and mutant cochlin in the cell, we performed immunocytochemistry in the presence of NP40, which permeabilises the cell membrane to allow diffusion of antibodies into the cell for detection of the protein intracellularly. We also performed immunostaining in the absence of cell permeabilising agents to determine whether there was detectable association of cochlins with cell surfaces or extracellular culture matrix. In the absence of NP40, no significant immunostaining was seen in the extracellular matrix of the transfected cells, even though cochlin is secreted in abundance as detected in the conditioned media by western blot analysis (discussed below). Perhaps in the unique extracellular environment of the inner ear, in the area of the cochlear and vestibular fibrocytes where cochlin is expressed endogenously, the protein shows stability in the matrix, as suggested by detection of abundant amounts of cochlin in the inner ear by western blotting, immunohistochemistry, and proteomic analysis.14,24 Furthermore, in the context of the inner ear extracellular matrix, cochlin may bind to, or interact with other components for stabilisation of the matrix. It is possible that the mutations in cochlin impair proper function of the protein in this context and result in cumulative deleterious effects over time through a dominant negative mechanism.
Secretion of normal and mutated cochlins
To assess intracellular cochlin levels as well as amounts secreted into the media, mammalian cultured cells were transfected separately with HA tagged wild type cochlin and the mutant variants (fig 3). The HA tag, relatively small in size, is placed at the C-terminus of the protein so as not to interfere with the signal secretion peptide. Cochlin levels in both the cellular fraction and conditioned media were separated by SDS polyacrylamide gel electrophoresis and analysed on western blots using an anti-HA antibody.
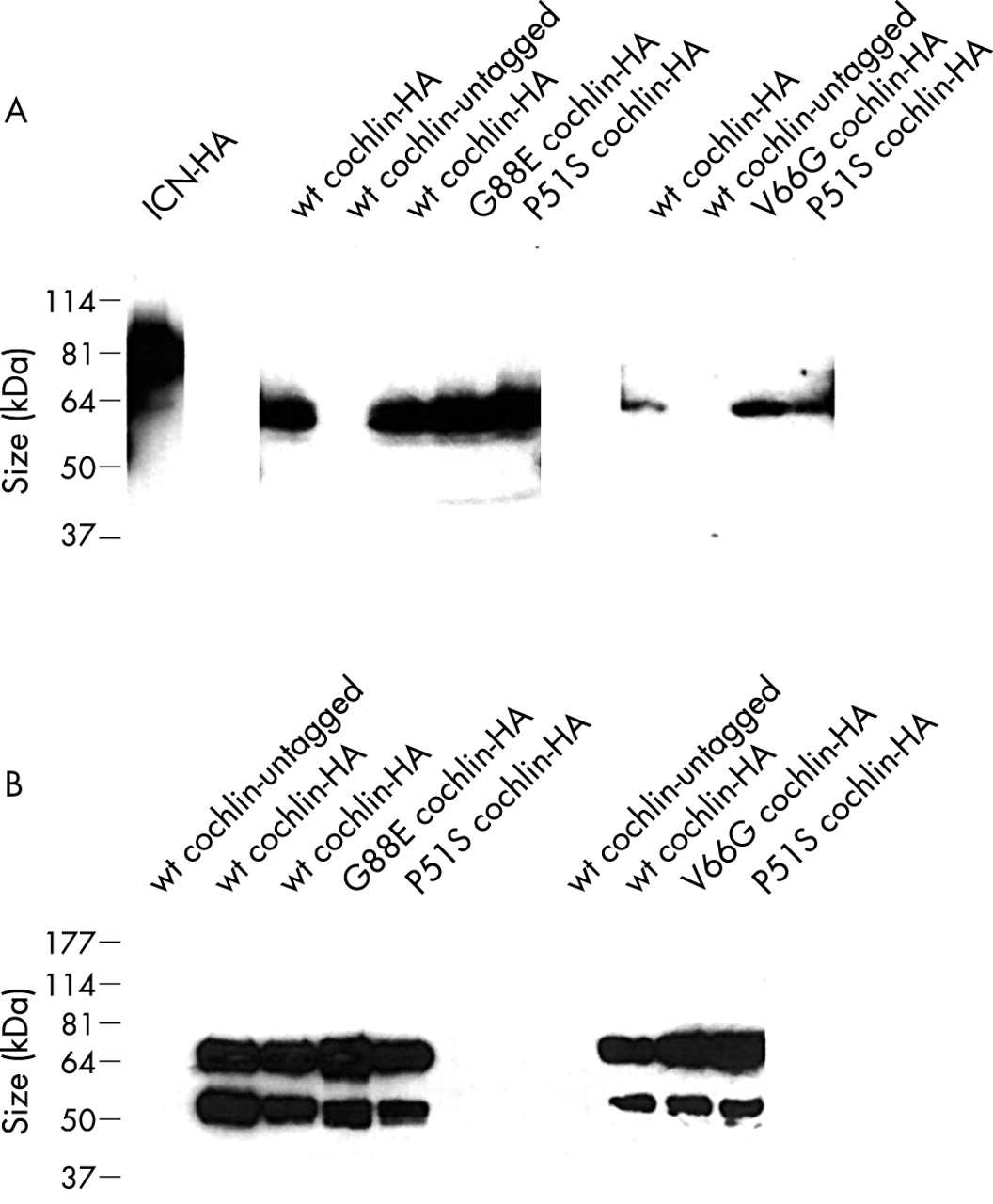
(A) Western blots of whole cell extracts of cultured 293T cells transiently transfected with HA tagged human COCH detected with anti-HA antibody. Similar intracellular cochlin protein levels (~60 kDa in size) are seen for wild type and mutant constructs (missense mutations, P51S, V66G, G88E, causing human deafness and vestibular dysfunction). An untagged COCH construct was used as a negative control for the western blots. An unrelated gene, the intracellular portion of NOTCH (ICN), also tagged with HA, was used as a positive control for transfections and western blotting. (B) Western blots showing secretion of cochlin by 293T cultured cells transiently transfected with HA tagged COCH constructs. The conditioned medium was immunoprecipitated with anti-HA covalently cross linked to sepharose beads, and proteins were solubilised from the beads by heating in SDS-PAGE loading buffer in the absence of β-ME. β-ME was subsequently added for electrophoresis on SDS-polyacrylamide gels for western blot analysis. A band of the expected size for full length cochlin is detected at equal levels for wild type cochlin and mutants known to cause human deafness and vestibular dysfunction (missense mutations P51S, V66G, G88E). A smaller ~50 kDa band, representing a processed product of cochlin, is also present in equal amounts for both wild type and mutant variants of cochlin. This band is absent in the untagged negative controls which were subjected to transfection and immunoprecipitation in the same manner as for the tagged samples; therefore, it does not represent a non-specific band or murine immunoglobulin heavy chain, which is covalently linked to the beads and is not expected to detach from the beads by solubilisation of proteins in SDS-PAGE loading buffer in the absence of β-ME. Note: the different panels represent independent experiments, reflecting parallel transfections, cell harvests, immunoprecipitations, and western blots. They are presented separately for internal comparison within the same experiment.
In cellular extracts of transfected cells, a ~60 kDa band representing full length cochlin was detected. To assess whether cochlin is a secreted protein, the media of transfected cells was subjected to immunoprecipitation with anti-HA antibody cross linked to sepharose beads, which concentrates and purifies HA tagged cochlin proteins. A prominent band corresponding to full length secreted cochlin as well as a smaller molecular weight band (~50 kDa) were detected in the HA tagged samples. Neither of these bands was present in untagged negative controls immunoprecipitated in parallel, indicating that the ~50 kDa band is not murine immunoglobulin heavy chain. Furthermore, the murine anti-HA antibody is covalently linked to sepharose beads and does not dissociate when proteins are solubilised in denaturing conditions in the absence of β-ME. It is therefore likely that the smaller sized ~50 kDa band represents a processed form of cochlin (discussed below in post-translational processing).
To determine whether missense mutations in cochlin result in either retention of the protein intracellularly or its degradation, levels of intracellular cochlin were evaluated on western blots of cellular lysates of cells transfected with equal amounts of wild type and mutant COCH constructs in parallel (fig 3A). A ~60 kDa band representing full length cochlin was seen in cellular extracts for the wild type and in the three mutants tested, with the same intensity in all transfected samples and within independent sets of transfections. These observations indicate that the presence of the three cochlin mutations does not result in their enhanced retention intracellularly, nor reduced stability leading to their removal or degradation. A possible caveat in interpreting these results is that during the time course of transient transfections, mutant proteins may not show effects of misfolding, but that over a longer time frame, stability and amounts of protein would show differences. It is also possible that the in vivo processing of the mutant proteins in the inner ear may be different from that in cultured cells, precluding detection by these experiments.
We also evaluated secreted levels of wild type and mutant cochlin proteins in media of transfected cells (fig 3B). Conditioned media from all samples were immunoprecipitated in parallel with anti-HA antibody and analysed on western blots. The secreted full length and processed cochlin bands were both detected for wild type and mutant cochlins, indicating that mutant variants appear to be secreted efficiently by the cell and are also subject to post-translational proteolytic processing. To ensure that protein levels were reflected accurately by immunoprecipitation and that the anti-HA beads were not limiting, cleared media were subjected to repeated immunoprecipitation. Western blot analysis of secondary immunoprecipitations showed equal, but much reduced, levels of cochlins across all samples.
These results indicate that the DFNA9 missense mutations analysed do not significantly impede secretion of cochlin in transiently expressing mammalian cells. Although previous data have shown misfolding of the bacterially expressed FCH/LCCL domain of cochlin in vitro as a result of the missense mutations,9 misfolding of this domain may not significantly affect the overall structure of the entire protein, or aberrant folding may be handled differently within transfected cells. If misfolding of the FCH/LCCL domain of cochlin does occur in the transfected cells, it does not appear to be sufficient to impair intracellular transport and secretion, or lead to aggregation or degradation of the protein. The observation of adequate secretion of the mutant proteins points towards events beyond secretion, in the extracellular matrix, where mutations in COCH may disrupt cochlin function or protein interactions.
Post-translational modifications of normal and mutated cochlins
Proteolytic cleavage
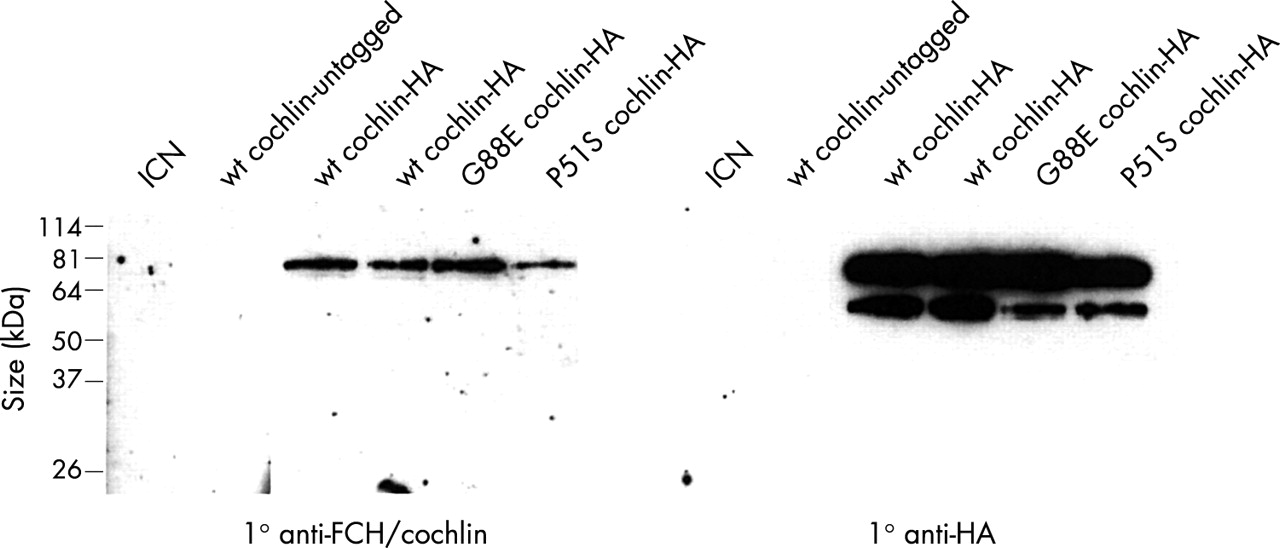
The smaller ~50 kDa form of cochlin in the secreted media of transfected cells was detected with an antibody to the C-terminal HA tag, suggesting that this isoform is created by cleavage at a site more proximal to the N-terminal end. To confirm this, we performed western blots with an antibody raised against a peptide in the FCH domain (fig 4). This antibody detected only the larger full length cochlin band, confirming that the ~50 kDa protein is derived from the C-terminal portion of cochlin. The ~50 kDa band was not present in the immunoprecipitated negative controls, indicating that it is not murine anti-HA immunoglobulin heavy chain, which is covalently linked to the agarose beads and therefore not dissociated from the beads in the absence of reducing agents.

Western blots of secreted HA tagged cochlin after immunoprecipitation with anti-HA antibody. The panel on the left was incubated with an anti-FCH/cochlin antibody raised against a small peptide in the N-terminal FCH domain of cochlin immediately C-terminal to the signal peptide. There is one band representing full length secreted cochlin. The smaller ~50 kDa processed cochlin product is not detected, indicating that it is lacking the region of the FCH domain against which the anti-FCH/cochlin antibody was generated, and that it is derived from the C-terminal portion of cochlin. The panel on the right was incubated with anti-HA antibody, and shows reactive bands of both full length and processed cochlin (as also shown in fig 3B). The HA epitope tags are at the C-terminal end of cochlin, confirming that the smaller band is derived from this end of the protein. Untagged cochlin, as well as intracellular NOTCH (ICN), which is not secreted into the media, serve as negative controls.
Previous reports have shown the presence of smaller isoforms of cochlin in cochlear extracts,14,24 suggesting this processing event occurs in vivo as well. Because whole cell extracts of transfected cells contain only the full length cochlin, whereas the media contains both the full length and the smaller processed form, post-translational cleavage probably occurs in a late secretory compartment. Interestingly, the apparent site of peptide cleavage is between the mutation bearing FCH domain of cochlin and the more C-terminal vWFA-like domains. Therefore, the ~50 kDa isoform of cochlin consists of only the vWFA-like domains and ivd2, and lacks the mutation bearing FCH domain. In light of this, it is important to study the fate of the ~10 kDa N-terminal cochlin fragment containing the FCH domain, predicted to be created by the cleavage that releases the ~50 kDa fragment, and to look for any potential differences in stability of this peptide between the wild type and the mutants. We introduced a FLAG epitope tag immediately C-terminal to the signal peptide with an intervening glycine residue between the tag and the FCH domain for appropriate exposure of the tag, and to provide some separation to allow for proper folding of the domain.
Western blot analysis of conditioned media immunoprecipitated with anti-FLAG showed the full length unprocessed cochlin, but not the ~10 kDa FCH domain polypeptide (not shown). Detection of the full length cochlin confirmed that insertion of the N-terminal FLAG tag had not interfered with secretion of the protein, and that the proteins had been successfully immunoprecipitated by the anti-FLAG antibody. Therefore, lack of detection of the ~10 kDa polypeptide suggests that it may have been subjected to further proteolysis and may not be a stable moiety. This is unexpected for the wild type cochlin, given the stability of the recombinant FCH fold in bacteria, and suggests that in mammalian cells this domain may be protected from proteolysis by interactions with the flanking vWF domains.
Glycosylation
Glycosylation is known to play an important role in the correct folding and assembly of peptides as a part of a complex set of mechanisms that provide quality control for protein synthesis and degradation.25 Glycosylated intermediates are immature proteins, allowing trafficking through different cellular compartments to reach their final destination or to function within the necessary developmental time frame or context. Incorrectly folded or assembled glycoproteins will not be exported from the ER or Golgi; instead, chaperones may be recruited to achieve correct functional conformation, or the misfolded intermediates may aggregate or be destroyed by the proteosome.15
In addition, many proteins are known to be glycosylated in their mature form. In particular, glycosylation is a very common phenomenon among transmembrane and secreted proteins. Glycoproteins serve very diverse biological functions depending on localisation, developmental context, and expression in different cell types, as well as in certain malignant states. Their functions vary widely and include roles in adhesion in cell-cell or cell-matrix interactions, or regulation of cell surface receptors and hormone functions, among many others.25
The amino acid sequence of cochlin has two consensus sites for asparagine linked (N linked) glycosylation; one is at amino acid residue 100 in the FCH/LCCL domain, and the other at amino acid residue 221 in the vWFA1 domain. To assess glycosylation of cochlin after transfection of eukaryotic cells with wild type and mutant COCH constructs, secreted cochlin proteins immunoprecipitated from the media were digested with endoglycosidases, Endo H and PNGase F. Both enzymes cleave N linked oligosaccharide groups, but the latter has a broader recognition of sugar groups,26 such that mature glycoproteins are PNGase F sensitive and Endo H resistant. Digestion of cochlin immunoprecipitated from the media of transfected cells with PNGase F results in a shift in electrophoretic mobility of the digested protein as compared to undigested and mock digested samples (fig 5). Western blots showed that the digested products migrate at a slightly lower molecular weight as compared to the undigested protein, indicating the presence of fairly small or few oligosaccharide groups, consistent with two predicted N linked glycosylation sites. PNGase treatment resulted in an electrophoretic mobility shift consistent with the removal of sugar residues, whereas Endo H produced no effect (not shown), as expected for a mature protein. Both bands shifted in this fashion, indicating at least one glycosylation site in the C-terminal portion of cochlin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Western blots of secreted HA tagged cochlin after immunoprecipitation and digestion with PNGase F endoglycosidase. Digested products show a shift in electrophoretic mobility as compared to undigested and mock digested samples. Digested products migrate at a slightly lower molecular weight, indicating the presence of fairly small or few oligosaccharide groups, consistent with two predicted N linked glycosylation sites in cochlin. Both full length and smaller processed forms of secreted cochlin show a shift in mobility after digestion. Secreted mutant cochlin proteins were also digested with PNGase F and also showed a shift in electrophoretic mobility similar to that seen with the wild type.
Although the known mutations in COCH do not change any amino acid residues that would alter the predicted N linked glycosylation sites in cochlin, it is possible that effects of mutations on the overall structure of the domain may interfere with oligosaccharide side chains. To assess whether cochlin missense mutations have any effect on glycosylation of the protein, the secreted mutant proteins were also digested with endoglycosidases in parallel with the wild type secreted protein (fig 5). The digested mutant proteins also showed a shift in electrophoretic mobility of both cochlin isoforms. Within the limits of detection by this method, there was no apparent difference in the glycosidase digestion of the mature secreted proteins between the wild type and mutant cochlin proteins tested, implying no obvious effects on glycosylation of cochlin owing to these mutations.
SUMMARY
We have shown that cochlin is a protein secreted via the endoplasmic reticulum/Golgi apparatus network, and is proteolytically processed and glycosylated in its mature form. Interestingly, we observed that the ~50 kDa processed form of cochlin was stable, whereas the ~10 kDa peptide representing the mutation bearing FCH domain could not be detected and may be subject to degradation in the absence of C-terminal vWFA domains. The proteolytic cleavage of cochlin seen in transfected cells, as well as previous data of smaller isoforms seen in vivo, also point out the interesting fact that there are forms of cochlin which lack the mutation bearing FCH domain. This observation, as well as the finding that mutant cochlins are not degraded by cellular quality control mechanisms, support the idea that the presence of the mutant protein may have a novel deleterious effect rather than cause pathology through haploinsufficiency.
Analysis of three missense mutations in cochlin responsible for DFNA9 deafness and vestibular disorder, in parallel with normal cochlin, showed that the mutants are able to be transported through the cells and secreted in a similar fashion as the wild type protein. They also undergo glycosylation and proteolytic processing. Overt degradation or aggregation, as often seen in the case of misfolded proteins, was not seen when mutated cochlin was expressed transiently in cultured cells. It is possible that over a longer time in vivo, a slow tendency to aggregate could manifest as cumulative damaging effects. This possibility is particularly relevant in light of the late onset and progressive nature of DFNA9, and the characteristic histopathological findings of abundant eosinophilic acellular deposits in the structures of the inner ear. In addition, the mutant protein, once secreted into the extracellular environment, may manifest pathology through its impaired function or altered interaction with other extracellular components of the cochlear and vestibular labyrinths, where cochlin is expressed abundantly.
Acknowledgments
We would like to thank Dr Elizabeth Sztul for helpful discussions. This work was supported by NIH/NIDCD grants DC03402 (to CCM) and CA82308 (to JCA).