Article Text

Abstract
In recent years, subtelomeric rearrangements have been identified as a major cause of mental retardation and/or malformation syndromes. So far, over 2500 subjects with mental retardation have been tested and reported of whom ∼ 5% appeared to have a subtelomeric rearrangement. In this review, the clinical aspects of each known (submicroscopic) subtelomeric deletion will be presented and the various methods available for detecting subtelomeric abnormalities will be discussed. Not only will the patients and their families benefit from a good collection and report of the various telomeric abnormalities and their clinical phenotype, but it will also give more insight into the aetiology of mental retardation and malformation syndromes.
- chromosome
- diagnostics
- submicroscopic deletion
- telomeres
Statistics from Altmetric.com
Mental retardation is a common handicap (2-3% of the general population) with an unknown cause in more than 50% of mentally retarded patients.1–4 Important causes are chromosome abnormalities which are detectable in 4-28% of cases, depending on the patient selection and techniques used.4,5 Deletions and/or translocations larger than 2-3 megabases (Mb) are mostly microscopically visible. 4p– (Wolf-Hirschhorn), 5p– (cri du chat), 9p–, 13q–, and 18p– syndromes are examples of microscopically visible deletions that mostly include the subtelomeric region and cause mental retardation associated with a specific phenotype.
For detecting submicroscopic subtelomeric abnormalities, Wilkie et al6 developed in 1993 a technique using hypervariable DNA polymorphisms. Two years later, Flint et al,7 using this method, identified previously undetectable abnormalities in 5% of 99 mentally retarded patients. This and other subsequent studies has led to the awareness that subtelomeric deletions below the level of the light microscope (<2-3 Mb) are a significant cause of malformation and mental retardation syndromes. In 1999, Knight et al8 reported a high rate of subtelomeric aberrations among children with moderate to severe mental retardation (IQ=50), whereas a lower yield was found in children with mild retardation (IQ 50-70) (7.4% versus 0.5%), thus again emphasising the importance of subtelomeric abnormalities in the former group of patients. Since then, several series of examinations of mentally retarded subjects, different in ascertainment, number of patients, and method used, have been reported (table 1). So far, over 2500 subjects have been tested and reported of whom ∼5% appeared to have a subtelomeric rearrangement. Compared to another well known condition causing mental retardation, namely the fragile X syndrome, subtelomeric deletions seem to be a more frequent cause of MR. The fragile X syndrome can be diagnosed in ∼1-2% of the mentally retarded.4,9,10 The relative high frequency of subtelomeric deletions should be interpreted with caution for various reasons. Firstly, the cases chosen for performing the telomere screen are probably selected for the so called chromosomal phenotype.11 Secondly, a reporting bias may influence the frequency as studies showing a low yield are less likely to be published. However, even if the frequency is somewhat lower than 5% it still will be a considerable step forward in diagnosing a significant number of mentally retarded subjects and counselling the families involved.
Studies of subtelomeres in patients with idiopathic mental retardation
The yield of new cases identified may even significantly increase by preselection based on family history and physical features. One important selective feature is the level of mental retardation; more subtelomere defects are found among the moderately to severely mentally retarded compared to the mildly retarded.8,12 However, subtelomeric abnormalities have also been described among mildly mentally retarded subjects. Based on the common features observed in a series of subtelomeric cases, a checklist was developed to facilitate preselection of cases for subtelomere testing,11,13 including (1) family history of mental retardation, (2) prenatal onset growth retardation, (3) postnatal growth abnormalities (either poor or overgrowth), (4) ≥2 facial dysmorphic features, (5) one or more non-facial dysmorphic features and/or congenital abnormality. Rio et al14 found congenital anomalies, behavioural problems, and postnatal growth retardation to be the most common features in their series, whereas Riegel et al15 reported the presence of more than one affected member in the family as the most important selection criterion in addition to the mental retardation combined with dysmorphic features, with or without major malformations and growth retardation.
CLINICAL STUDIES
Mental retardation is the key feature in patients with subtelomeric defects. Some of the submicroscopic subtelomere deletions result in a specific phenotype which may direct the clinician towards the diagnosis. In these patients, FISH analysis of a single and specific subtelomere will be sufficient to confirm the diagnosis. However, the majority of cases with subtelomeric defects lack a characteristic phenotype, so far. For these cases a general subtelomere screen is required to achieve a diagnosis. For this group effective clinical preselection is essential because of the technical complexities and cost of screening for telomere deletions (see discussion).
In the following sections the submicroscopic subtelomeric deletions and their clinical presentation are described for each chromosome end.
Fig 1 shows clinical photographs of various patients with subtelomere deletions.
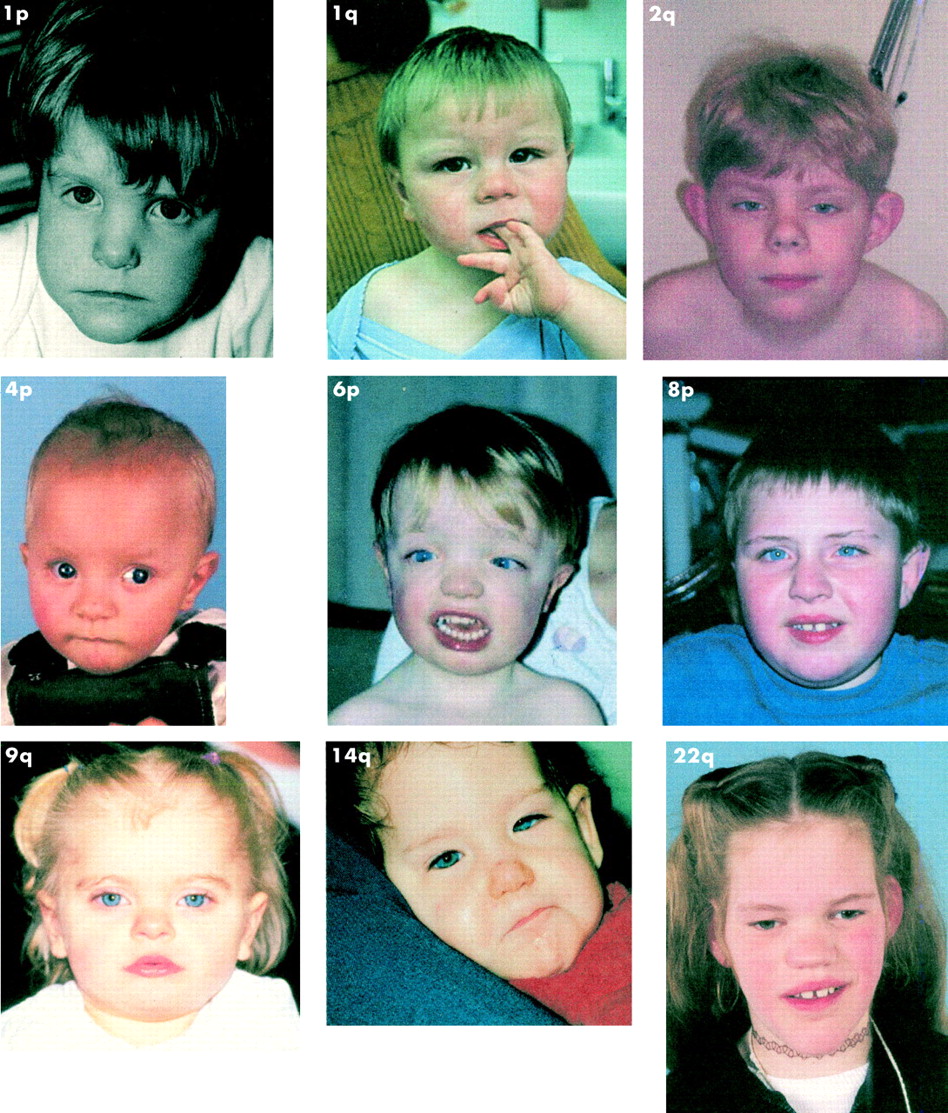
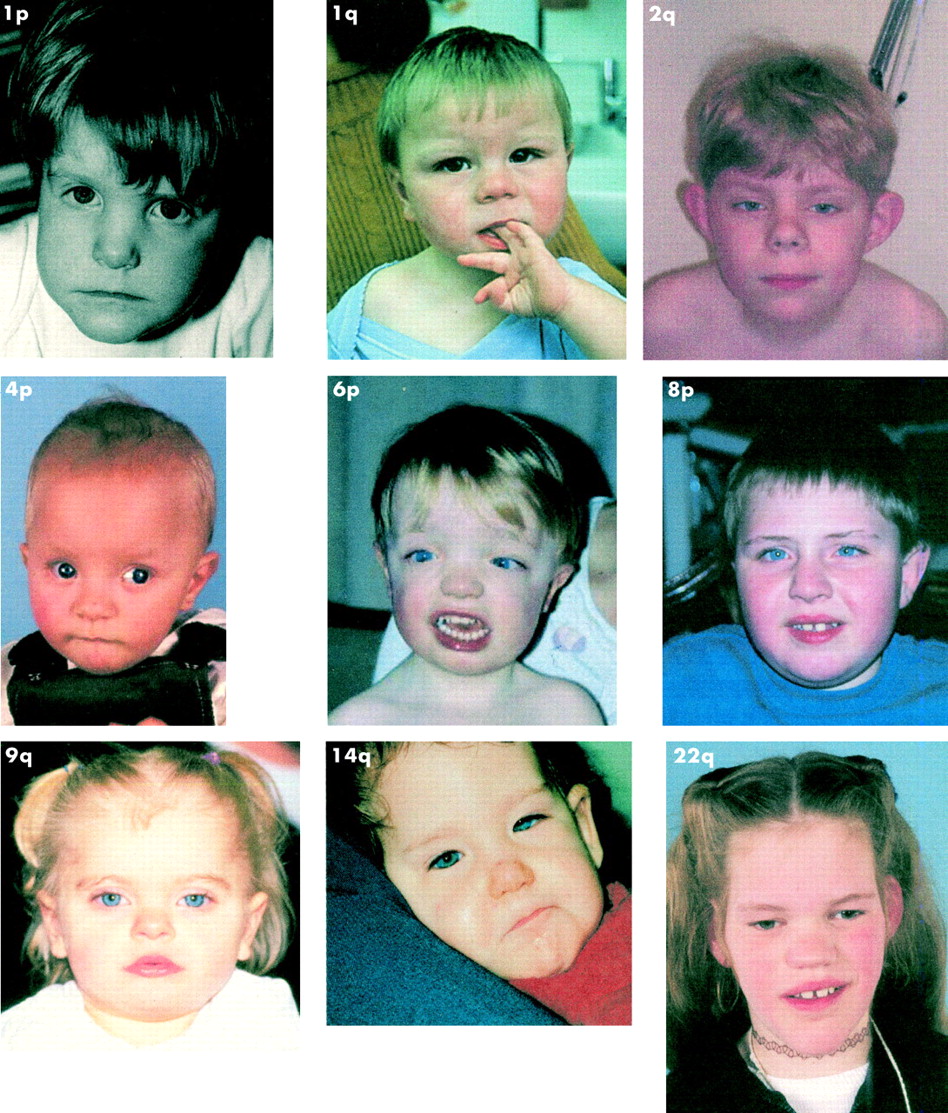{kind=link}
Clinical photographs of various patients with subtelomere deletions.
1p
Shapira et al16 reported on clinical and molecular aspects of 1p36 deletion in 14 patients in 1997. Subsequently, Slavotinek et al17 reviewed 39 patients with pure 1p36 monosomy, the majority with a microscopically visible deletion, and confirmed the association with hypotonia, mental retardation (usually severe), growth abnormalities (growth retardation, microcephaly, obesity), and facial dysmorphism with a large anterior fontanelle, prominent forehead, deep set eyes, depressed nasal bridge, and midface hypoplasia, ear asymmetry, a pointed chin, and orofacial clefting (10- 40% of patients). In some cases, the straight and low set eyebrows are striking and can be helpful in the diagnosis. Variable cardiac malformations, cardiomyopathy, seizures, ventricular dilatation, sensorineural hearing loss, and visual problems have been reported. The Ebstein heart anomaly, which, in general, is very rare in chromosome aberrations, has been observed in at least three cases with 1p36.3 deletion.14,18,19 Disturbed behaviour has been reported varying from temper outbursts to self-injurious behaviour.
Although a putative neuroblastoma tumour suppressor gene has been mapped to the 1p36.1-1p36.3 region, in the series of Wu et al20 none of the patients (n=30, ages 2-14 years) developed neuroblastoma. They also showed that the breakpoints were highly variable within 1p36 and that 21 out of 27 de novo deletions were maternally derived. Using five highly polymorphic minisatellite probes for 1p36.3, Giraudeau et al21 found three 1p– patients among 567 mentally retarded subjects and Rio et al14 found three patients among 150 severely mentally retarded patients, making it one of the more common cryptic subtelomeric deletions.
1q
De Vries et al22 reported two unrelated boys with a submicroscopic terminal 1q44 deletion and growth and mental retardation, severe microcephaly, hypospadias, corpus callosum abnormalities, cardiac abnormalities, gastro-oesophageal reflux, and characteristic facies. Facially, they had short noses with a long, smooth philtrum, a thin upper lip, and full round facies with periorbital fullness. One case had a de novo 1q44-qter deletion and the other der(1)t(1;13)(q44;q34) caused by a balanced maternal t(1;13). In the same family, a female fetus (17 weeks) with microretrognathia and a large midline cleft in addition to facial dysmorphism was found to have the same 1q monosomy and 13q trisomy as her brother. The authors suggested the location of gene(s) involved in normal midline development in the subtelomeric region of 1q. Rossi et al12 reported another boy who was profoundly mentally retarded with pachygyria, seizures, facial dysmorphism (not further specified), scoliosis, and toe syndactyly caused by a der(1)t(1q;12p).
A profoundly mentally retarded boy with severe microcephaly, cleft palate, facial anomalies (upward slanting eyebrows, small palpebral fissures), postaxial polydactyly of the left hand, brachydactyly, and generalised amyotrophy and a der(1)t(1;18)(q44;p11.3)mat was reported by Riegel et al.15
Baker et al23 reported a 15 year old male with borderline IQ, short stature but normal head circumference, and facial dysmorphism (long face, almond shaped eyes with upward slanting palpebral fissures and thick eyebrows, broad nasal base with fleshy nares, smooth philtrum, and thin upper lip), short distal phalanges, and cryptorchidism, and a deletion of 1qter and a duplication of 1pter probably resulting from a large parental pericentric inversion. The mother’s chromosomes were normal and the father was not available for testing. Another case with severe mental handicap, pre- and postnatal growth retardation, microcephaly, ptosis and ophthalmoplegia, and adducted thumbs was reported in the series of Rio et al.14
2p
Riegel et al15 reported a 2 year old severely mentally retarded boy with severe microcephaly, bilateral cleft lip and palate, and seizures. A female fetus (sib) was terminated with microcephaly and bilateral cleft lip and palate. Both had a der(2)(t(2;7)(p25.2;q36.1).
2q
Phelan et al24,25 reported four cases with apparent Albright hereditary osteodystrophy (AHO) and del(2)(37.2) detected by high resolution chromosome analysis. Wilson et al26 reported five other patients with brachymesophalangism and mental retardation; four had cytogenetically visible de novo deletions of chromosome 2q37, and one had a microdeletion at 2q37. Since then several other patients with an AHO-like phenotype and a submicroscopic 2q37.3 microdeletion have been reported, some caused by a familial submicroscopic translocation, notably t(2;8)(q37.3;q24.3) and t(2;17) (q37.3;17q?).27,28 This AHO-like phenotype consists of mental retardation, short stature, round face, brachymesophalangism, and epilepsy. Behaviour is generally friendly; however, hyperkinesia, aggression, self-mutilation, or psychiatric problems can be present.27 Ghaffari et al29 reported a 6 month old boy with craniofacial dysmorphism somewhat suggestive of Noonan syndrome, tetralogy of Fallot, inguinal hernia, and a partial monosomy 2qter and trisomy 17qter detected by comparative genomic hybridisation (CGH). A der(2)t(2;7)(q73;q36) was detected in a severely cognitively retarded 3 year old boy with facial dysmorphism (prominent forehead, downward slanting palpebral fissures, arched eyebrows, long eyelashes, small mouth with short philtrum and thin upper lip), a few café au lait spots, pectus carinatum, and talipes equinovarus.30
Mildly mentally retarded patients with 2qter deletions and an inconsistent clinical phenotype have been reported31–33 andseveral patients even had a phenotypically normal parent with a similar deletion suggestive of a familial polymorphism.31 It seems that the 2qter deletion polymorphism is the most frequent of all subtelomeric polymorphisms. Ballif et al34 reported eight cases (5%) in their series of 154 patients with the 2q telomeric polymorphism also found in a healthy parent.
In the series of 150 severely mentally retarded patients reported by Rio et al,14 three cases have been reported.
3p
Over 25 cases with 3p– syndrome and deletion breakpoints at 3p25-p26 have been reported.35 However, all were microscopically visible except for two sibs who were initially reported by Verloes et al36 as the autosomal recessive GOMBO syndrome (acronym for Growth retardation-Ocular abnormalities-Microcephaly-Brachydactyly-Oligophrenia). The phenotype was caused by a 3pter monosomy and 22qter trisomy as a result of der(3),t(3;22)(p25;q13).37
Characteristic features of the 3p– syndrome include low birth weight, microcephaly, mental and growth retardation, trigonocephaly, hypotonia, ptosis, telecanthus, downward slanting palpebral fissures, and micrognathia. Other less frequent abnormalities are preauricular pits, cleft palate, postaxial polydactyly, heart defects, and renal and gastrointestinal anomalies.
The majority are de novo but familial cases have been reported.37–39 Remarkably, Knight et al39 reported a phenotypically normal mother and child both with a terminal 3p25.3 deletion, which suggests that a deletion distal to 3p25.3 does not need to have an apparent deleterious effect.
Unlike 4p– and 5p–, where 80% of de novo deletions arise in the paternal germline, 3p deletion can either be of paternal or maternal origin.
Large deletions may include the von Hippel-Lindau (VHL) disease gene so screening for VHL in patients with large deletions should be considered.40 The ATP2B2 gene (PMCA2) centromeric from VHL has been considered a candidate for congenital heart malformations.41 However, Green et al35 narrowed down the critical region for heart defects using five cases and concluded that ATP2B2 is outside this region.
Haploinsufficiency of the CALL gene at 3p26.1 that codes for a member of the L1 gene family of neural cell adhesion molecules has been considered to be responsible for a part of the mental impairment.42 Higgins et al43 mapped a locus to chromosome 3p25-pter responsible for an autosomal recessive type of non-syndromic mental retardation in a large highly inbred family.
3q
Only one submicroscopic 3qter deletion (de novo) has been reported so far. This 1 year old moderately mentally retarded boy had facial dysmorphism (not further specified), horseshoe kidney, and hypospadias.12
A limited number of microscopically visible 3qter deletion patients (n=5) have been reported with growth and mental retardation, hypotonia, and ear abnormalities in common. Three out five patients died before the age of 2 years.44
4p
Terminal deletions of 4p are well known for their characteristic phenotype of Wolf-Hirschhorn syndrome.45 The Wolf-Hirschhorn syndrome critical region (WHSCR) is located in 4p16.3 and in approximately 25% of the patients with WHS the (terminal) deletion comprising this region is only detectable by FISH. Deletions with a size less than 3.5 Mb have been described46 that result in a distinct but relatively mild WHS phenotype without malformations. This mild variant of WHS is also known as Pitt-Rogers-Danks syndrome with growth retardation, microcephaly, mental retardation, seizures, and a distinctive facial appearance. Anderlid et al,32 in their series of patients screened for subtelomeric deletions, described a 3 year old girl with severe mental retardation, no language, epilepsy, short stature, hypertelorism, downward slanting palpebral fissures, broad forehead, and low set ears with a deletion that was not detectable by high resolution chromosome analysis, but included WHSCR. The critical deletion region has been narrowed down to 165 kb at 4p16.3.47 Subtelomeric deletions of only 100-300 kb from the telomeric end do not result in the WHS phenotype.
Prenatal diagnosis of a fetus with a cryptic t(4;18)(p15.32;p11.21) was reported by Kohlschmidt et al.48
The Lambotte syndrome (microcephaly, holoprosencephaly, intrauterine growth retardation, facial anomalies, and early lethality) initially reported as an autosomal recessive disorder49 was found to be caused by a deletion of 4p16.2-pter and a duplication of 2q37.1-qter.50
4q
A few cases with a submicroscopic 4q35-qter deletion have been reported albeit with a limited clinical description. Two patients from one family had an additional partial trisomy of 20p owing to a familial t(4;20)(q35;p13) and they presented with mild mental retardation, growth retardation, facial dysmorphism (flat philtrum, wide mouth, and low set ears), heart defects (VSD), and bilateral vesicoureteric reflux.29 A similar family (or the same family) with similar clinical features, except for the severity of the mental retardation that was severe in the affected patients, was reported by Knight et al.8
Another case was partially trisomic for 6qter owing to a familial t(4;6)(q35;q27) and he had facial dysmorphism (unspecified in the report), syndactyly, lymphatic dysplasia, and a micropenis with hypospadias.8 A 5 year old mildly mentally retarded boy with an oval shaped face, deep set eyes, and left ptosis (also found in father) with a der(4)(t(4;10) (q35.2;q26.3)mat was reported by Riegel et al.15 Two mentally retarded half sibs both with minor facial anomalies, heart defects, and a distal tracheal stenosis and a cryptic del 4q34-qter and dup 12p13-pter with a common balanced carrier mother were reported by Fritz et al.51
5p
The clinical picture of distal deletions of 5p is known as cri du chat syndrome.52 The most typical feature of this syndrome, the cat-like cry, is the result of a subtelomeric deletion located more distal than the deletions that cause the facial features and severe developmental delay in cri du chat syndrome.53–55 However, in the series of Cerruti Mainardi et al52 the patients with the most distal deletions only had speech delay and no cat-like cry. Rossi et al12 described a cryptic 5p deletion resulting from a t(4p;5p) in a 15 year old child with moderate mental retardation, triangular face, gingival hypertrophy, prominent incisors, posteriorly angulated ears, and behavioural disturbances.
5q
Two cases with a submicroscopic 5q35.3-qter deletion have been reported so far.23 One was a 6 year old, moderately delayed female with an additional trisomy of 16qter owing to a familial t(5;16) and mild dysmorphic features (dense hair, prominent forehead, large mouth, thin upper lip, and thick lower lip) and normal growth. The same group reported another unrelated 12 year old female with a similar deletion 5qter and duplication 16qter, but this time de novo, and a low-normal IQ (80-89), normal growth, mild facial dysmorphism (mild epicanthic folds, upward slanting palpebral fissures, low nasal bridge, short philtrum), slender hands and feet, cardiac abnormalities (mild pulmonary valve stenosis and large VSD), and vesicoureteric reflux.
One microscopically visible de novo terminal deletion of 5q35.3 has been reported in a mildly delayed 15 month old boy with macrocephaly, mild retrognathia, anteverted nares with low nasal bridge, telecanthus, minor ear lobe anomalies, bell shaped chest, diastasis recti, and brachydactyly.56 Two cases with a small deletion involving 5q35.1-q53.3 have been reported: a markedly delayed 9 month old girl with macrocephaly, epicanthic folds, downward slanting palpebral fissures, broad nasal bridge, anteverted nares, micrognathia, bifid uvula, nuchal redundancy, brachysyndactyly, clenched fingers, and seizures,57 and a 7½12 year old girl with subtle facial anomalies and a complex heart disease including ventricular myocardial non-compaction.58
6p
One 4 month old moderately mentally retarded boy with a de novo submicroscopic 6p25-pter deletion was included in the study of De Vries et al.11 He had a low/normal growth, hypertelorism, long palpebral fissures, flat malar region, large mouth, small fifth fingers, long/broad halluces, scoliosis, cryptorchidism, congenital heart defects (small VSD, PDA, bicuspid aortic valve, and coarctation of the aorta), and hypermetropia with a posterior embryotoxon (M Splitt, personal communication). The microscopically visible 6p25-pter has been associated with anterior chamber eye defects (corneal opacities/iris coloboma/Rieger anomaly), hypertelorism, downward slanting palpebral fissures, tented mouth, smooth philtrum, palatal malformations (high arched and/or cleft palate), ear anomalies, deafness, abdominal hernia, small external genitalia, and cardiac defects.59–60
6q
In the series of Knight et al,8 a 10 year old mildly retarded boy with macrocephaly and a submicroscopic 6q27-qter and a partial trisomy 12p13.3-pter owing to a familial t(6;12)(q27;p13.3) was included. Except for the macrocephaly and some minor facial dysmorphisms (thin upper lip and downturned corners of the mouth), no abnormalities were present (C McKeown, personal communication). The macrocephaly and thin upper lip might also be the consequence of the partial trisomy 12pter.61 Batanian et al62 reported a 5 year old mentally retarded boy with a half cryptic translocation 6qter and 2pter (partial trisomy 2p25.3-pter and monosomy 6qter, the der(6) was microscopically visible) with minor dysmorphic features (epicanthic folds, thin upper lip, flat philtrum, and low set, large ears), which supports the relatively mild phenotypic presentation of submicroscopic 6qter deletions. However, Rossi et al12 reported a 1 year old child with congenital chylothorax, facial dysmorphism (not further specified), and absence of sacral vertebral fusion caused by a familial der(6)t(6q;16p). Two severely retarded uncles with facial dysmorphism and short stature had a similar der(6). Colleaux et al63 reported a severely mentally retarded boy with a 6qter deletion and a 10qter trisomy with a long and thin face, microstomia, cleft lip and palate, dental anomalies, tall stature, and seizures. The same patient was also reported by Joly et al.33 Moreover, Anderlid et al32 also reported three cases with severe mental retardation showing that various levels of retardation are associated with 6qter deletions. One 4 year old, severely mentally retarded boy had a de novo 6qter deletion and microcephaly, bilateral syndactyly of the second and third toes, inguinal hernia, skeletal abnormalities (vertebrae and ribs), epilepsy, and facial dysmorphism (high, broad nasal bridge, long eyelashes, large tented mouth, and large ears). Two severely mentally retarded sibs with a deletion 6qter and a duplication 6pter owing to a paternal pericentric inversion 6 had similar dysmorphic characteristic features: hypoplastic midface, high forehead, blepharophimosis, short, pointed nose, thin upper lip and high palate, wide supratentorial ventricles and heterotopias, and severe feeding problems.
Larger and microscopically visible 6qter deletions (6q25-qter) have been associated with microcephaly, retinal abnormalities, cleft palate, facial dysmorphism (notably ear anomalies, broad nasal tip, high nasal bridge, epicanthic folds, and long philtrum), limb anomalies, genital hypoplasia, agenesis of the corpus callosum, and cardiac defects in addition to a moderate to severe mental retardation.64–66
7p
Joly et al33 reported a severely retarded 1 month old girl with a deletion 7pter and partial trisomy for 16qter with dolichocephaly, hypertelorism, micrognathia, vertebral anomalies (not specified), anteriorly placed anus, and brain stem dysfunction. Eleven cases with microscopically visible deletion 7p22.1-pter have been reported, some with facial anomalies (notably broad, flat nasal bridge, low set, malformed ears), digital anomalies, cardiac defects, and hypoplastic genitals.67
7q
The 7q36.1-qter region contains two important genes, the HLXB9 gene involved with Currarino syndrome (sacral dysgenesis, anorectal atresia, and a presacral mass)68 and the Sonic hedgehog (SHH) gene involved with holoprosencephaly (HPE3).69 Brackley et al70 reported a 1½12 year old, severely developmentally delayed girl with a submicroscopic der(7)t(2;7)(q37;q36) and a low birth weight and hypotonia. There was marked dysmorphism: microcephaly, hypertelorism, upward slanting palpebral fissures, left microphthalmos and right anophthalmos, posteriorly rotated ears, and downturned corners of the mouth. The neck was short and there was fixed left talipes. The family history was positive for stillborn children, miscarriages, and neonatal deaths. A familial t(2;7)(q37;q36) was detected.
Wang et al71 reported two sibs with sacral agenesis, anorectal malformation, microcephaly, and marked developmental delay. One also had a cleft palate and recurrent urinary tract infections. Both had a submicroscopic 7q36-qter deletion owing to a familial t(7;12)(q36;q24). They also reported a 10 year old girl with sacral dysgenesis and moderate retardation, microcephaly, low weight and height, and hypotelorism. She had a 7qter deletion owing to a maternal t(7;8).
8p
Microscopically visible distal 8p deletions have been associated with growth and mental impairment, minor facial anomalies, congenital heart defects, and behavioural problems. De Vries et al72 reported two mildly mentally retarded, non-dysmorphic cousins with behavioural problems including inappropriate sexual behaviour and pyromania and a terminal submicroscopic 8p deletion caused by a familial t(8;20)(p23;p13). The frequently observed microcephaly in patients with microscopically visible deletions of 8pter was lacking in both cousins, suggesting that the gene(s) causing the microcephaly were centromeric to the deleted region. The absence of cardiac defects in the cousins confirmed the more proximal location of gene(s) causing these abnormalities and supports involvement of the GATA4 gene in other reported cases with microscopically visible 8pter deletions. Moreover, the current cases predict the presence of a putative gene(s) involved in behaviour in the most telomeric 5.1 Mb of the short arm of chromosome 8.
In their series of 70 patients with mental retardation, Sismani et al73 found a 5 year old moderately mentally retarded girl with a subtle 8p deletion (in retrospect also detectable by high resolution banded chromosome analysis) and microcephaly, obesity, hypotonia, and speech delay.
Another severely mentally retarded boy with facial dysmorphism (not further specified) was reported in the series of 150 mentally retarded patients by Rio et al.14 However, cases with a microscopically visible 8p23.1-pter and normal IQs have also been reported.74–76
8q
No submicroscopic deletions of 8q24.3-qter have been reported.
9p
The clinical features of the 9p– deletion syndrome include dysmorphic facial features (trigonocephaly, upward slanting palpebral fissures, and a long philtrum) and mental retardation. The majority of the patients have a microscopically visible deletion with a breakpoint in 9p22. However, the trigonocephaly, upward slanting palpebral fissures, and long philtrum can also be seen in patients with a smaller deletion (9p24, own observation). Upward slanting palpebral fissures and mental retardation, but no trigonocephaly or long philtrum were found in a girl with a cryptic translocation that resulted in a deletion 9p24.77
Deletions of 9p have been associated with 46,XY gonadal dysgenesis and the smallest region of overlap has been mapped to the tip of chromosome 9 (9p24.3).78,79 This region involved in male to female sex reversal has a high gene density and several candidate genes for sex reversal or gonadal dysgenesis have been identified. However, so far no mutations have been found in these genes.
9q
In the series of Knight et al,8 two severely mentally retarded sisters (6 and 11 years old) with a der(9)t(9;13)(q34;p11.1) were reported. Both were severely hypotonic with similar dysmorphism: brachymicrocephaly, coarse facies, long eyebrows with synophrys, large tongue, upturned nose with prominent nares, tented mouth, short philtrum, and low set, posteriorly rotated ears (O Quarrell, personal communication). Both sisters had periventricular white matter changes, epilepsy, joint laxity, and sensorineural deafness. The youngest also had a congenital heart abnormality (PDA and VSD). The mother was a carrier of the balanced t(9;13). Rossi et al12 reported a de novo 9qter deletion in a moderately mentally retarded child with facial dysmorphism (not further specified), and also Anderlid et al32 in a 25 year old severely mentally retarded woman with epilepsy, synophrys, hypertelorism, and strabismus, and Rio et al14 in a severely mentally retarded female with obesity, abnormal genitalia, and hyperactivity.
10p
One case with a der(10)t(10;12)(p14;p13.2)pat has been reported, a boy with mental retardation, cleft palate, 2-3 cutaneous syndactyly of the toes, and cryptorchidism.80 Joly et al33 reported a 2 month old, severely mentally retarded infant with low birth weight, hypotonia, severe myopia, and a perineal angioma and der(10)t(10;12)(p12;p12) diagnosed by comparative genomic hybridisation. In retrospect, the derivative chromosome 10 was detectable in the original karyotype.
However, larger and visible deletions of 10p13-pter have frequently been reported (n>25). These are characterised by frontal bossing, short, downward slanting palpebral fissures, ear anomalies, micrognathia, congenital heart defect, vesicoureteric abnormalities, and developmental delay.81,82 As the clinical presentation of the latter deletion is relatively mild, it could be possible that the smaller subtelomeric deletions do not cause an abnormal phenotype or have only minor abnormalities.
In the 10p13/14 region, the GATA3 gene associated with HDR (hypoparathyroidism, deafness, renal dysplasia) was identified83 and a critical region for the second DiGeorge syndrome locus (DGCR2) was localised proximal to this locus.84BRUNOL3 has been suggested as a candidate gene for the thymus hypoplasia/aplasia in partial monosomy 10p patients.85
10q
A severely mentally retarded 32 year old woman with short stature, microcephaly, and non-specific facial dysmorphism and a cryptic deletion of 10qter and duplication 20qter was reported by Ghaffari et al.29 The same chromosomal abnormality was detected in her moderately mentally retarded cousin.
A 2 year old moderately mentally retarded boy with a der(10)t(10q;16p) has been reported with facial dysmorphism (not otherwise specified) and a hypoplastic penis.12 Colleaux et al63 reported two sibs with severe mental retardation, enophthalmia, long nose, full lips with everted lower lip, foot deformation (not further specified), and autism. An 8 year old severely mentally retarded girl with a 10qter monosomy and a 16qter trisomy with low birth weight, microcephaly, high nasal bridge, thin upper lip, small chin, and strabismus has been reported.33,63
Two moderately mentally retarded sibs with a der(10)t(4;10)(q35.2;q26.3)mat were reported with a triangular face, hypertelorism, downward slanting palpebral fissures, short, upturned nose with broad and prominent root and hypoplastic alae, thin upper lip, downturned corners of the mouth, bilateral preauricular tags, smooth palmar creases, and short fingers.15 One had in addition a bifid uvula.
A 10q telomere deletion was reported by Martin et al86 in a 22 month old developmentally delayed boy with pachygyria, seizures, cerebellar hypoplasia, absent corpus callosum, ichthyosis, mild arthrogryposis and dysmorphic features including sparse scalp hair, absent eyebrows, and short upper limbs. However, his phenotypically normal mother carried the same telomere deletion.
Patients with microscopically visible 10q26.1-qter deletion (n=20) seem to have a consistent phenotype including mental disability, growth retardation (pre- and/or postnatal), with microcephaly, triangular face, hypertelorism, strabismus, prominent nasal bridge, low set ears, micrognathia, short neck, cryptorchidism, ano/genital defects, and cardiac and renal anomalies.87–89
11p
Only one patient with a submicroscopic 11pter deletion has been reported. This 6 year old severely mentally retarded child had epilepsy, West syndrome, metabolic acidosis, microcephaly, ogival palate, simplified ears, thick lips, and micrognathia.12 Remarkably, there are no reports of microscopically visible 11pter deletions except for one infant with a complex chromosomal rearrangement involving chromosomes 11, 13, and 21,90 either suggesting that these deletions are probably lethal or simply do not occur.
11q
The distal 11q deletion or Jacobsen syndrome is caused by a microscopically visible deletion of the chromosomal bands 11q23, q24, and/or q25. Commonly observed features of this syndrome included mild to moderate mental retardation, postnatal growth retardation, trigonocephaly, facial dysmorphism (hypertelorism, epicanthus, ptosis, upward slanting palpebral fissures, short nose/long philtrum, retrognathia, high arched palate), cardiac defects, digit/hand/foot anomalies, and thrombocytopenia/pancytopenia. Many patients with a terminal 11q deletion have thrombocytopenia or pancytopenia and this seems to be related to the absence of band 11q24. Deletions distal to 11q24.1 do not produce the typical 11q– syndrome.91 Chromosome band 11q23 is probably associated with craniosynostosis and cardiac defects whereas bands 11q24 and 11q25 are associated with facial dysmorphism and thrombocytopenia.92 Terminal deletions extending proximal to 11q23.3 are probably lethal.
Recently, we saw a newborn boy with a congenital heart defect and thrombocytopenia. Routine karyotyping was normal. Because of the clinical features, FISH with a subtelomeric 11q probe was performed and showed a de novo deletion. A cryptic 11q;18q translocation that resulted in two cousins with a deletion 11q and with a deletion 18q was described by Schultz et al.93 The break in chromosome 11 was within the subtelomeric region (a 11q subtelomeric probe hybridised on both translocation chromosomes). An adult woman with the derivative chromosome 11 had mild mental retardation, short stature, and no dysmorphic features.
Clarkson et al31 reported an 18 year old woman with an 11q24.3-11qter deletion and a duplication of 11p15.5-11pter owing to a paternal pericentric inversion. She had features reminiscent of Jacobsen syndrome and a head circumference on the 97th centile consistent with Beckwith-Wiedemann.
12p
A 12 year old girl and her 8 year old brother, both with a submicroscopic deletion of 12p13.3-pter and a partial trisomy 6q27 owing to a maternal t(6;12)(q27;p13.3), were included in the series of Knight et al.8 The girl was mildly mentally retarded with pre- and postnatal growth retardation including microcephaly, high palate and late tooth eruption, and prominent tip of the nose. Her brother was moderately mentally retarded and had like his sister pre- and postnatal growth retardation, high arched palate, and late tooth eruption. However, he had more dysmorphic features including downturned corners of the mouth, anteverted nares, long, flat philtrum, thin upper lip, clinodactyly, widely spaced nipples, and delayed bone age (C McKeown, personal communication). Growth retardation and microcephaly might also be caused by the 6qter abnormality as this has also been described in a boy with partial trisomy 6q26-qter.94
Moreover, a 15 year old, moderately disabled boy and his mother with (unspecified) learning difficulties were reported by Baker et al23,95 with a 12pter deletion. He had normal growth and minimal dysmorphic features (prominent ears and deep set eyes), persistence of some primary dentition, short neck, mild thoracic kyphoscoliosis, right sided aortic arch, and aggressive behaviour. Two genes in the region, SLC6A12, a betaine/GABA neurotransmitter transporter gene expressed in liver, heart, skeletal muscle, and placenta with widespread distribution in the brain, and WNT5B from the Wnt family, which encodes secreted growth factor-like proteins, have been suggested as candidates for the mental retardation, facial dysmorphism, and digital and dental anomalies.95
12q
Submicroscopic deletions of 12qter have not been reported.
13q
Kleefstra et al96 reported two sisters with a submicroscopic 13q34-qter deletion and partial trisomy for 8q24.3-qter owing to a maternal t(8;13)(q24.3;q34). The two mildly mentally retarded sisters (aged 31 and 25 years) had a similar phenotype consisting of obesity, skin atrophy of the lower limbs, mild facial dysmorphism (thin lips, bulbous nose with large nares, and a wide columella), and muscle weakness resulting in lordosis, valgus position of the knees, and pes planus.
In the series of De Vries et al,11 two cases with a submicroscopic 13qter deletion were included. One 17 year old severely mentally retarded girl had a submicroscopic 13q34-qter deletion and a partial trisomy 1q44-qter. She had a normal birth weight, was macrocephalic at 18 years (OFC >98th centile) with short stature (<3rd centile), and had various dysmorphic features including small overfolded ears, microphthalmia, low posterior hairline, small mouth with a high palate, short fingers with clinodactyly, and small 5th toes (Homfray, personal communication). The other was a moderately mentally retarded, 2½12 year old boy with a de novo submicroscopic 13q34-qter deletion with low birth weight, severe microcephaly, and mild facial dysmorphism (hypertelorism).
Riegel et al15 reported a moderately retarded, 4 year old boy with severe microcephaly, oval shaped face, epicanthic folds, small, thick ears, micrognathia, short fingers, brachymesophalangy V, short feet with large toes, and abnormal palmar creases.
A 5 year old, severely mentally retarded boy with microcephaly, facial dysmorphism (high forehead, flat occiput, hypertelorism, epicanthic folds, short nose with bulbous tip, flat philtrum, open mouth with downturned corners), fifth finger camptodactyly, and talipes valgus with absence of the fifth toe nails, and a 46,XY,der(13)t(4;13)(p16.3;q34)pat was reported by Helias-Rodzewicz et al.30 His severely mentally retarded father’s sister had similar clinical features and the same chromosomal abnormalities.
14q
One boy with developmental delay, microcephaly, reflux/vomiting, a long face with flat supraorbital ridges, ptosis, blepharophimosis, wide nasal root, long beaked nose, two hair whorls, a small mouth, a pointed chin, a narrow chest with low widely spaced nipples, single palmar crease, camptodactyly, and bilateral hearing loss was presented by Lunt et al (P Lunt, personal communication). There was a positive family history; two previous children of the mother from different fathers died with MCA (a girl at the age of 5 weeks and a male fetus at 26 weeks’ gestation). The mother had a submicroscopic t(14;17)(q32.3;p13.3).
Baker et al23 reported a 17 year old, moderately mentally retarded male with pre- and postnatal growth retardation and microcephaly, facial dysmorphism (low set simple ears, mild facial asymmetry, hypotelorism, blepharophimosis, broad prominent nose, high arched palate), slender hands, and absence of the distal interphalangeal creases of the third and fourth fingers.
Several microscopically visible 14qter deletions have been reported mostly as a result of a ring chromosome 14. Like the submicroscopic case, these patients showed microcephaly, narrow elongated face, blepharophimosis, flat nasal bridge, low set ears, and micrognathia. However, retinal dystrophy and seizures seem to be restricted to patients with ring chromosomes. Similar 14q– features were observed in a boy with an apparently balanced translocation between 14q and 21p, who was shown to have a submicroscopic terminal deletion of 14q32.3 with FISH.97 Van Karnebeek et al98 reported another example of a submicroscopic 14q32.31-qter in mildly mentally retarded girl. These cases suggest that the characteristic 14qter phenotype is caused by a deletion of 14q32.3-qter and that blepharophimosis in combination with features such as microcephaly are strong indicators to perform 14qter FISH analysis.
15q
No submicroscopic deletions of 15q26.3-qter have been reported.
Cases with a ring chromosome 15 have de novo deletions of the distal part of 15q (q26.3, q26.2, and/or q26.1) and are characterised by growth and mental retardation, microcephaly, triangular face, hypertelorism, high arched palate, abnormal ears, micrognathia, and brachydactyly.99 A minority have cardiac and/or urogenital abnormalities. Depending on the size of the deletion, these patients may be missing one copy of the insulin-like growth factor receptor gene (IGF1R) and some do have features suggestive of Russell-Silver syndrome100–101
16p
The combination haemoglobin H disease and mental retardation was first reported by Weatherall et al in 1981.102 Wilkie et al103 showed in 1990 that this α-thalassaemia/mental retardation syndrome was caused by a terminal (submicroscopic) deletion of 16p13.3. Besides the α-thalassaemia/mental retardation, the associated dysmorphic features are mild: hypertelorism, downward slanting palpebral fissures, broad, flat nasal bridge, and epicanthic folds. Holinski-Feder et al104 reported 10 affected family members with ATR-16 syndrome caused by a subtelomeric submicroscopic der(16)t(3;16)(q29;p13.3). They showed mild facial dysmorphism similar to previously described cases and a low birth weight, hypotonia, pes equinovarus, and undescended testes in males. Warburton et al105 reported two sisters with a submicroscopic subtelomeric deletion of 16p13.3-pter and a partial trisomy of 1p36.33-pter. The oldest presented with a left corneal dermoid cyst and a unilateral iris coloboma at birth with preauricular skin tags and a patent ductus arteriosus, all suggestive of Goldenhar syndrome. At 15 years of age she was small (height, weight, and OFC <3rd centile) with an unusual face, including bitemporal narrowing with sloping forehead, very broad almost bifid nose, marked hypertelorism with epicanthic folds, slightly upward slanting palpebral fissures, small preauricular tags, open mouth appearance, and dental malocclusion. The extremities showed long, slim fingers, 5th finger clinodactyly, and ulnar deviation of the middle fingers. Her younger sister showed a more or less similar phenotype. Eussen et al106 reported a boy with a submicroscopic deletion of 16p13.3-pter and duplication of 8q24.3-qter with tuberous sclerosis, adult polycystic kidney disease, and hypomelanosis of Ito. Brown et al107 reported a newborn with cranial tubers and subcortical renal cysts suggestive of tuberous sclerosis and additional atypical features such as bilateral nasal colobomas of the eyes, inguinal hernias, glandular hypospadias, cryptorchidism, and facial dysmorphism (telecanthus, short palpebral fissures, broad nasal tip, small mouth with thin lips and small chin) caused by a der(16)t(16;19)(p13.3;p13.3).
The SOX8 gene, 700 kb from the telomeric end, has been suggested as a good candidate gene for the mental retardation.108 Notably, Horsley et al109 studied 21 independent deletions from the 356 kb telomeric region of 16p13.3 and did not find any discernible phenotype other than α-thalassaemia. This suggests that the additional phenotypic effects are caused by larger deletions of 16p13.3 (>1 Mb).
16q
Only one submicroscopic deletion of 16qter has been reported which was caused by a 16q;19p rearrangement. This 11 year old child had moderate mental retardation, facial dysmorphism (not further specified), precocious puberty, short stature, hypernatraemia, and behavioural problems.12 Werner at al110 reported a 5 year old boy with a small distal interstitial deletion of chromosome 16 (del(16)(q23.1q24.2)) and bilateral coloboma of the iris, short stature, moderate developmental delay, and a few facial anomalies (broad forehead, bushy eyebrows, and a large nose and mouth). A locus for cataract has been suggested within 1 Mb of 16qter based on findings in patients with ring 16.111
17p
Terminal deletions of 17p result in a distinctive phenotype, Miller-Dieker syndrome, which is characterised by severe mental retardation, lissencephaly, cerebral atrophy, agenesis of the corpus callosum, microcephaly, and kidney anomalies. It has been known for some years that microdeletions can also cause this syndrome and the critical region is within the terminal band 17p13.3. Several families with a cryptic translocation and Miller-Dieker syndrome have been described.112–114 The lissencephaly in Miller-Dieker syndrome is the result of deletion of the LIS1 gene. Isolated lissencephaly has been described in patients with a balanced translocation with a breakpoint in the LIS1 gene.115 Mutchinick et al116 described an 8 year old girl with mental retardation, postnatal growth deficiency, hypotonia, seizures, microcephaly, cortical atrophy, partial agenesis of the corpus callosum, facial anomalies, pectus excavatum, long fingers, and bilateral talipes equinovarus with a deletion telomeric to LIS1. However, phenotypically normal subjects with telomeric 17p deletions up to 600 kb have been reported.86
17q
One de novo 17qter deletion has been reported in a 15 year old child with moderate mental retardation, cardiopathy (not further specified), and extreme thinness12 and another 17qter deletion and 12qter duplication, owing to a paternal translocation, in a mildly mentally retarded girl with pre- and postnatal growth retardation, ataxia, autistic features, cleft palate, ventricular septal defect, congenital nystagmus, and facial anomalies (not further specified) and a partial agenesis of the corpus callosum. As the growth factor receptor bound protein 2 located on 17q25.1 has been suggested as a candidate locus for Silver-Russell in two translocation patients,117 this gene might also be involved in causing the extreme thinness in the former single case
No further submicroscopic deletions of 17q25-qter have been reported so far.
18p
Although numerous cases with microscopically visible 18pter deletions have been reported, none of them is a submicroscopic deletion of 18p11.3-qter. The deletion 18pter patients have short stature with variable facial dysmorphism such as a round face, hypertelorism, flat nasal bridge, wide mouth with downturned corners, and a single maxillary incisor. Notably, 10% of cases have holoprosencephaly for which the putative HPE4 gene has been mapped to 18p11.3,118 and Gripp et al119 reported four missense mutations in the TGIF gene in 268 patients with HPE.
Remarkably, a 37 year old female with a (visible) deletion 18p11.2-pter and a partial trisomy for 2p25-pter was reported who had normal intelligence, suggesting that such small 18pter deletions are only associated with mental retardation if (some degree of) holoprosencephaly is present.120 Familial microscopically visible 18p deletions have been reported121–123 and some patients with normal or borderline intelligence have been observed.
18q
The 18q– or de Grouchy syndrome is characterised by mental retardation, hearing loss (most often resulting from narrow or atretic auditory canals), midfacial hypoplasia, growth deficiency, and various limb anomalies (for example, proximally placed thumbs, tapering fingers and fifth finger clinodactyly, coxa valgus, and abnormal toe position). Most patients have deletions in 18q21. However, more distal deletions (18q23) may also result in the classical phenotype, but there exists a wide phenotypic variability.124 Slavotinek et al125 reported a severely mentally retarded 15 year old girl with a de novo submicroscopic 18q23-qter deletion and growth retardation, hypotonia, seizures, and cerebral atrophy. She had severe myopia with bilateral choroidoretinal atrophy, midfacial hypoplasia, and a narrow mouth with downturned corners consistent with 18q deletion syndrome. Knight et al8 reported in their series a boy with a de novo der(18)t(X;18)(q28;q23) who was severely mentally retarded with growth retardation (including microcephaly), crowded midface, small carp-like mouth, maxillary hypoplasia, small, widely spaced nipples, and a small hypoplastic scrotum (R Winter, personal communication).
Vogels et al126 described a family with a cryptic translocation t(5;18)(qter;qter) that had resulted in unbalanced offspring with features characteristic of the 18q deletion syndrome (growth deficiency, nystagmus, narrow auditory canals, genital hypoplasia, behavioural problems) and features that are observed in 5q duplication (umbilical and inguinal hernias and congenital heart defect). Another familial cryptic translocation (11q;18q) was described by Schultz et al.93 Two adult men in this family had a subtelomeric deletion 18q and duplication 11q. They were marginally mentally retarded without dysmorphic features.
19p
No submicroscopic deletions of 19p13-pter have been reported. In 1984 only one patient with a visible partial deletion of 19p was reported with a prenatal onset of growth retardation (including microcephaly), hypertelorism, flat nose, micrognathia, low set ears, downturned corners of the mouth, high palate, umbilical hernia, non-specific deafness, punched-out lesions of the retina, and red cell abnormalities.127 However, the deletion of 19pter in the latter case is questionable because of the poor quality of the chromosome analysis.
19q
No submicroscopic deletions of 19q13.4-qter have been reported. Microdeletions in 19q13.2 have been associated with mental retardation, skeletal malformations, and Diamond-Blackfan anaemia.128–130 This suggests a contiguous gene syndrome which includes the ribosomal protein S19 that is mutated in Diamond-Blackfan anaemia.131
20p
Baker et al23 reported a 10 year old moderately mentally retarded male with a submicroscopic deletion of 20pter and microcephaly, facial dysmorphism (long face, deep set eyes with upward slanting palpebral fissures, and a small mouth with a short philtrum), and flat feet. He had epilepsy from 7 years of age and delayed secondary dentition. His mentally retarded mother had the similar 20pter deletion with similar facial characteristics. Unfortunately, other family members were not available for testing.23
20q
No submicroscopic deletions of 20q13.3-qter have been reported.
21q
No submicroscopic deletions of 21q22.3-qter have been reported. Small visible deletions of 21q22.3 and ring chromosome 21 have been associated with holoprosencephaly.132,133
22q
Most reported cases with a 22q13.3 deletion have been microscopically visible but since the development of submicroscopic screening methods of the telomeres, more than 10 cases with a submicroscopic or cryptic deletion have been reported.
In addition to the developmental delay, monosomy 22q13.3 has been associated with other clinical features: hypotonia, severe expressive language delay leading to absence of speech, pervasive behaviour, and subtle facial dysmorphism. The facial features do not seem to form a characteristic pattern, although the majority of the microscopically visible cases do have dolichocephaly, ptosis, epicanthic folds, and dysplastic ears. For the few submicroscopic 22q13.3 deletion cases, the facial features are even more subtle and variable.
Precht et al134 reported two cases with some similarities with Angelman syndrome. However, De Vries et al135 found no evidence for 22q13.3 -qter deletions in 44 cases with features suggestive of Angelman syndrome but without the characteristic 15q abnormalities.
One interesting case with general overgrowth and features suggestive of FG syndrome has been reported.136 Anderlid et al32 reported a mildly mentally retarded 34 year old woman with probably a very small de novo 22qter deletion and autistic disturbance, epilepsy, ataxic gait, and discrete facial findings. Another severely mentally retarded girl with a de novo 22qter deletion and 20qter duplication was reported with facial dysmorphism, simian creases, a thin corpus callosum, and abnormal white matter.32
ProSAP2 (the human homologue of the proline rich synapse associated protein 2) has been suggested to be causative for the 22qter deletion syndrome after identification of a disruption of this gene in a patient with a balanced t(12;22) and the 22qter phenotype.137
Xp
Numerous patients with deletions of Xpter have been reported and the clinical presentation is dependent on the deleted genes and can for males result in a combination of the following disorders: short stature (SS), X linked recessive chondrodysplasia punctata (CDPX), mental retardation (MRX), ichthyosis (XLI), and Kallmann syndrome (KAL).138,139 One gene for short stature, the short stature homeobox containing gene (SHOX),140 was also shown to be mutated or deleted in families with Leri-Weill dyschondrosteosis.141,142
Although a putative locus for mental retardation (MRX49) has been located distal to the STS locus,143 Tobias et al144 reported a boy with a normal intelligence and a telomeric deletion including the STS locus as the result of a der(X)t(X;Y)(p22.31;q11.21). Clinically there is a much similarity with the case reported by De Vries et al145 with a submicroscopic Xp22.31-pter deletion with a breakpoint just upstream of the STS locus. The latter boy however was mentally retarded.
Xq
No submicroscopic deletions of Xq28-qter have been reported. Microscopically visible Xqter deletions are associated with ovarian failure in females.146
DISCUSSION
The identification of subtelomeric rearrangements as a cause of mental retardation has made a considerable contribution to diagnosing patients with mental retardation and counselling of the families involved. The reported frequency of subtelomeric deletions of ∼5%, which is a considerable proportion of cases with mental retardation of unknown cause, might actually be a little overestimated, as in the major studies published so far selection of cases has probably occurred. This selection has been based on the “chromosomal phenotype” as has also been shown by several groups.11,14,15,30 Such selection has been required for some time being the currently available techniques, either FISH with telomere specific probes or molecular analysis based on polymorphic markers, are labour intensive. This is likely to change when new techniques will allow for larger number of patients to be tested.
Currently, two techniques are commonly used for detecting subtelomeric abnormalities (reviewed by Knight and Flint147). First is the use of polymorphic microsatellite markers localised in the subtelomeric region.63,148 A disadvantage of this technique is that for detecting hemizygosity for a certain marker, DNA samples of the parents are required. Another limitation is that most of the informative markers are located a relative large distance from the telomere and therefore small subtelomeric deletions can easily be missed using this method. However, with this technique isodisomy can be detected.
The second method is FISH of probes (BAC, PAC, or P1 clones) localised in the subtelomeric region to metaphase chromosomes. The initial problem of cross hybridisation, which hampered this method for certain subtelomeres, has largely been overcome. This second generation set of telomere specific BAC, PAC, and P1 clones are within 500 kb from each telomere and are therefore suitable to detect small subtelomeric rearrangements.149
Currently, all the subtelomeres can be tested on a single chromosome metaphase slide with a device developed by Knight et al,8 the Cytocell Ltd Multiprobe technique. However, this latter technique is still labour intensive. Therefore, new techniques have been developed to overcome the limitations of these commonly used techniques. Armour et al150 developed the multiplex amplifiable probe hybridisation (MAPH) methodology which allows assessment of copy number at specific genetic loci. This technique has also been proven to work for screening of subtelomeric chromosome abnormalities.73 Another promising technique is multiplex ligation dependent probe amplification (MLPA) which unlike MAPH does not require immobilisation of sample nucleic acids with additional washings.151 Veltman et al152 reported an array based comparative genomic hybridisation (CGH) to detect subtelomeric chromosome rearrangements. This technique is shown to be a rapid and sensitive automated procedure, but it requires an array facility. If the set of clones is extended over other regions of the genome, the array CGH will eventually allow for a “whole genome screen”. Using a 400 microsatellite marker panel, Rosenberg et al153 showed that a genome wide microsatellite scan can be used to detect submicroscopic chromosomal aberrations. Extension to thousands of markers equally divided over the genome will make such a scan very sensitive for detection of cryptic chromosomal abnormalities, but, at the moment, costly as well. Although multiplex FISH (M-FISH) allows for the detection of cryptic abnormalities as well,154–156 it is (or is likely to be) less sensitive than a microsatellite scan or a microarray CGH.
All these new techniques will also allow for detection of other as yet unknown submicroscopic interstitial deletions in the genome. The yield in diagnosing new chromosomal abnormalities related to mental retardation is likely to be considerable.
The increasing number of very small chromosomal aberrations that will be found in the near future will confront the clinician with various problems. When should a submicroscopic deletion be considered to be the reason for the mental retardation? If the microdeletion has been observed in other patients with mental retardation either within the same family or in unrelated cases, the deletion could mostly be regarded as causative. However, even microscopically visible deletions exist which do not cause mental retardation in all probands, for example, the deletion of the entire short arm of chromosome 18. Another problem is the (telomeric) polymorphisms, for example, the 2q subtelomeric region, which might be more common than previously considered.34 When other clinical features are part of the clinical presentation in a single patient, then comparison between other patients with similar deletions will be required. This will be essential for counselling the parents and family involved. For some of the common forms of subtelomeric deletions such as 4p, 5p, and 9p (table 2), the phenotype is quite consistent. However, for most of the subtelomeric deletions the number of patients with similar deletions reported is still limited or no cases have been reported at all. When new techniques become available, even more new microdeletions will be detected which will be at first single cases only.
Frequency of specific submicroscopic subtelomere deletions
It is remarkable that for certain subtelomere regions no deletions have been reported so far, such as 8q, 12q, 18p, 19p, 19q, 20q, and 21q (table 2). Because of the relatively novelty of the technique, it is conceivable that such deletions have just not yet been found simply because they are rare, but will certainly be found in the future. Another explanation is that these deletions are not associated with a “characteristic” chromosomal phenotype, for instance lacking the mental retardation, and therefore are simply not looked for in the right patient population. Maybe certain deletions are lethal, but that does not explain why certain large microscopically visible deletions involving the subtelomeric region are reported whereas submicroscopic ones have not been found, for example, 18p and 19p. Finally, some subtelomeric deletions may just not occur because of stability of the specific subtelomeric chromosome region. The future will tell us which of the above explanations/mechanisms are involved in these rare subtelomeric rearrangements.
In almost half of the patients, the telomeric deletion appeared to be de novo.8 It is likely that the whole genome is vulnerable to similar, albeit interstitial, microdeletions. Of course there are some major differences. First, a de novo telomeric deletion requires a single chromosomal breakpoint in contrast to the double break with interstitial deletions and might therefore occur more frequently. Secondly, the subtelomeric microdeletions are more likely to give a phenotypic effect, most commonly involving mental retardation, because of the gene richness of these regions.157 Moreover, one of the well known interstitial microdeletions, the 22q11.2 deletion, is in a considerable proportion of cases not even associated with mental retardation. However, the majority of the known interstitial microdeletions do have mental retardation as the major clinical presentation. So far these interstitial microdeletions have been found because of their characteristic phenotype. The majority of patients with subtelomeric deletions have just been diagnosed by using the telomere screening method with limited clinical guidance, the chromosomal phenotype. After identification of similar telomeric deletions, clinical resemblance between patients has been sought and found,17,136,158 although sometimes the number of patients is too limited (yet) to identify a phenotype. If facilities to identify interstitial microdeletions are in place then it is likely that large numbers of new deletions will be found. Like for the subtelomeres, we will have to collect similar cases in order to identify the clinical presentation which will allow proper genetic counselling of the family. As most newly identified interstitial deletions will be single cases, this knowledge will not be easy to obtain. An adequate collection of clinical data of those rare cases will be required in order to help the clinician and the family to understand the meaning of these findings in their patients and their relatives. Therefore, a collaborative European consortium has started to facilitate the collection and subsequently the distribution of this knowledge with EU funding (ECARUCA, www.ecaruca.net). Such a collection will not only help patients and their families and clinicians but will also give more insight into molecular mechanisms involved in the aetiology of mental retardation and malformation syndromes.
Acknowledgments
We wish to thank Drs Kets, Newbury-Ecob, Flinter, Lynch, and Bongers for kindly providing clinical photographs. B B A de Vries was supported by a grant from ZON-MW (The Netherlands). The photograph of the 1p– patient has been published before18 and is reprinted with the permission of Wiley-Liss Inc, a subsidiary of John Wiley & Sons Inc.
REFERENCES
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵