Article Text

Abstract
The hepatorenal fibrocystic (HRFC) syndromes are a heterogeneous group of severe monogenic conditions that may be detected before birth. Commonly, HRFC syndromes present in the neonatal and paediatric age, with consistent developmental abnormalities mostly involving the liver and kidney. The changes include the proliferation and dilatation of epithelial ducts in these tissues with abnormal deposition of extracellular matrix. In this review, we examine the clinical features and differential diagnoses of this group of syndromes, including autosomal recessive polycystic kidney disease (ARPKD), juvenile nephronophthisis (NPHP), Meckel-Gruber syndrome (MKS), Bardet-Biedl syndrome (BBS), and Jeune asphyxiating thoracic dystrophy (JATD). Extrahepatic manifestations include mostly bone and central nervous system abnormalities, dysmorphic features, and developmental delay. Previously, it has been suggested that ARPKD, JATD, and Ellis-van Creveld syndrome (EvC) may arise from defects in differentiation in a common developmental pathway. We review recent molecular advances in the recessive HRFC syndromes and discuss this hypothesis.
- fibrocystic
- kidney
- liver
- recessive
Statistics from Altmetric.com
The congenital hepatorenal fibrocystic syndromes are a group of severe, mostly autosomal recessive, monogenic disorders that are characterised by a common pathological appearance, with the presentation of multiple defects in the liver and kidney as the most predominant feature. In the liver, increased hepatic fibrosis often associates with cysts lined with biliary epithelium and a variable degree of intrahepatic biliary tract dilatation. Cystic lesions also affect the kidneys and their severity determines the clinical presentation and long term prognosis for many HRFC syndromes. It has been suggested previously that hepatic and renal malformations in ARPKD, JATD, and EvC result from defects in developmental pathways shared by many organ systems.1–5 In this review we provide a brief update on the molecular pathology and genetics of these disorders. We examine how recent molecular genetic advances in the characterisation of the ARPKD and NPHP genes provide insights into the “common developmental pathway” hypothesis for the aetiology of these clinically heterogeneous group of disorders. The molecular basis of a number of rarer HRFC syndromes remains unknown, and it is hoped that these insights might provide a rationale for the selection of candidate genes in the future, on the basis of protein function.
PATHOLOGY OF AUTOSOMAL RECESSIVE FIBROCYSTIC DISEASES
HRFC disease is characterised by changes in the parenchymal tissues of the liver, kidney, and sometimes pancreas or other organs. They include the proliferation and dilatation of epithelial ducts, and proliferative changes in the extracellular matrix of stromal connective tissue. One of the manifestations that has received intensive study are related to the ductal plate malformation of the liver and comprise proliferation and dilatation of the intrahepatic bile ductules, with a variable degree of fibrosis and cyst formation.6,7
The intrahepatic bile ducts develop out of sheets of primitive hepatic epithelial cells in a pattern determined by the branching of the portal vein and surrounding mesenchyme.6,8 The primitive epithelial cells that are in direct contact with the portal vein mesenchyme transform into bile duct type cells by forming a single layer and later two layers (fig 1A). Development progresses by the formation of a cleft between the two layers of cells known as the ductal plate (at gestational weeks 9 to 12). In response to unknown developmental signals, the biliary cells migrate from the ductal plate into the portal mesenchyme, with biliary structures still in partial contact with the ductal plate (the remodelling ductal plate stage at gestational weeks 13 to 17, fig 1A). Finally, remodelled bile ducts are formed that are centrally located in the portal tracts, without contact with ductal plate remnants (the remodelled bile duct stage at gestational weeks 18 to 40, fig 1A). The unincorporated epithelium of the ductal plate then disappears. Any interruption in the remodelling of the ductal plate can result in the persistence of embryonic duct structures, termed the ductal plate malformation (fig 1B). The initial lesion in the liver is thought to be excessive ductular proliferation, characterised by the formation of multiple dilated periportal biliary ductules, presumably resulting from a defect in complex epithelial-mesenchymal interactions. The ductal plate malformation is a histopathological characteristic of a number of fibrocystic syndromes, such as MKS (fig 2A, B), hepatic-pancreato-renal or Ivemark syndrome (HPRS), and JATD1–5 (table 1). The ductal plate malformation can also result in subsequent liver damage, and inflammation can induce the formation of fibrosis, and in some cases, cirrhosis.6,9
Summary of the clinical details of monogenic fibrocystic syndromes, listing hepatic, renal, and other associated anomalies

Embryogenesis of bile ducts. (A) Schematic diagrams of normal embryogenesis, progressing from the ductal plate stage (gestational weeks 9 to 12) to remodelled bile ducts of the portal tract (gestational weeks 18 to 40). Remodelling is thought to be the result of epithelial-mesenchyme inductive interactions. The embryonic structures are in cross section, with a branch of the portal vein (lumen in white) and a cuff of surrounding mesenchyme (dense red dots) at the central axis. The red lines represent the ductal plate. Refer to the text for further details. (B) Examples of biliary dysgenesis, resulting in ductal plate malformations owing to incomplete remodelling. The ductal plate (red lines) is in the form of either an interrupted circle or peripheral tubular structures. The ductal plate malformation is a characteristic manifestation of a number of congenital fibrocystic syndromes that are inherited in an autosomal recessive manner.
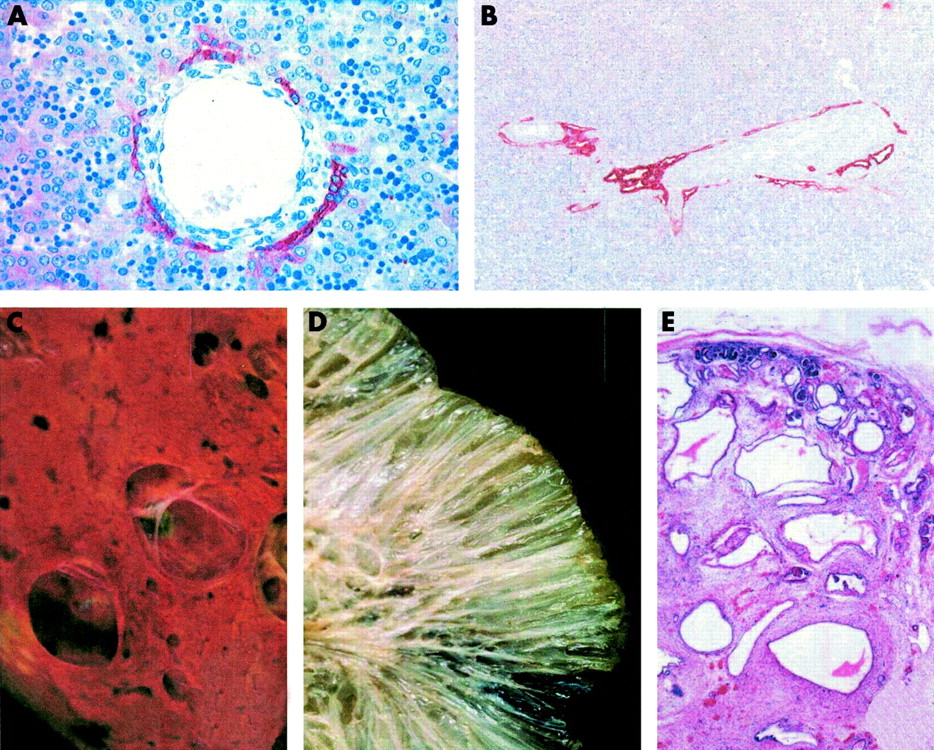
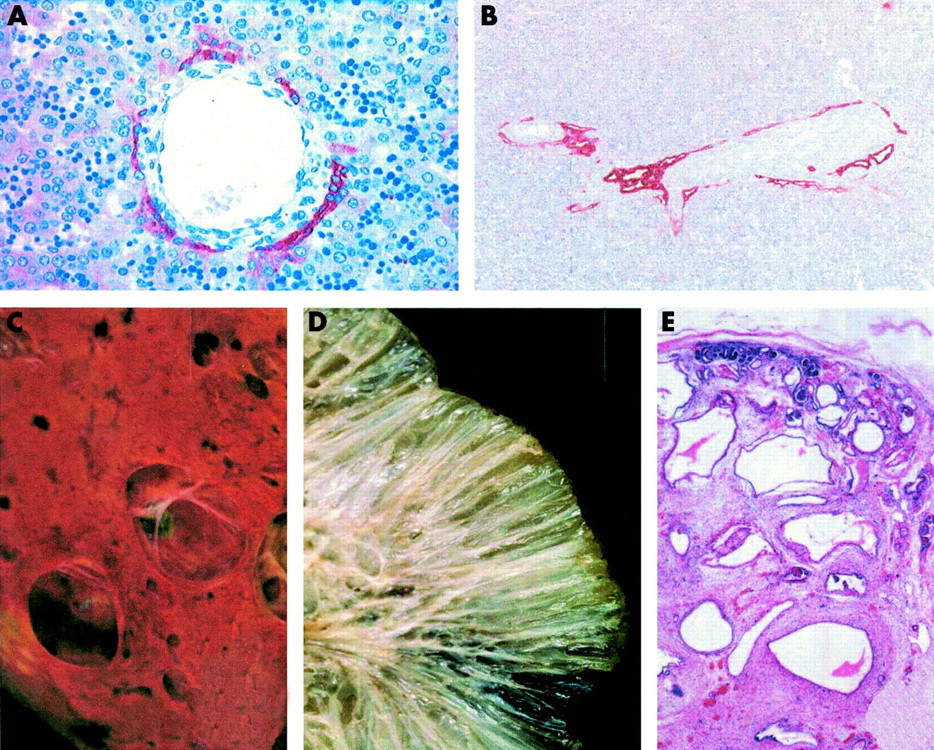
Examples of the ductal plate malformation and cystic changes in the liver and kidney for a range of congenital fibrocystic disorders. Panels (A) and (B) show immunohistochemical detection of a mixture of cytokeratin epitopes to visualise ductal and bile duct cells. (A) Normal control, showing a portal vein branch surrounded by connective tissue and cells expressing cytokeratin forming a discontinuous, partially double layered ductal plate. (B) A ductal plate malformation (type I). Two mid to large sized portal tracts are shown that are close to the hilum hepatic and with central portal veins. The portal tracts show a cystic dilatation of the primitive biliary structures that are located at the limiting plate. There is a moderate increase in intraportal connective tissue between the endothelium of the portal vein and the ductal epithelium. Panels (C) to (E) show examples of cystic changes in the liver and kidney for a range of fibrocystic disorders. (C) Liver cysts or polycystic liver disease of an adult affected with autosomal dominant polycystic kidney disease (ADPKD). Cysts in ADPKD can be derived from any segment of the nephrons, from glomerular capsule to collecting ducts. The cysts can be numerous to countless, fluid or blood filled, and range in size from millimetres to centimetres in diameter. (D) The cut surface of a kidney from a child affected with autosomal recessive polycystic kidney disease (ARPKD). The kidney shows a sponge-like appearance, with dilated elongated channels at right angles to the cortical surface. The dilated elongated channels almost completely replace the medulla and cortex. (E) Diffuse cystic kidney dysplasia of a fetus (2nd trimester of pregnancy) affected with Meckel-Gruber syndrome, showing cysts throughout the cortex and the medulla. There are small and medium sized thin walled cysts that vary greatly in size. The lobar organisation of the kidneys is preserved but corticomedullary differentiation is mostly absent.
Hepatic fibrosis may manifest either as a consequence of ductal plate malformation within a monogenic fibrocystic disorder, in response to hepatocyte damage and subsequent necrosis, or as necrosis independent hyperstimulation of matrix producing cells. In the liver, the major cellular sources of extracellular matrix are activated Ito or hepatic stellate cells. These are fat storing cells that resemble fibroblasts, and in response to chronic hepatocyte damage they differentiate into myofibroblasts. These cells then secrete matrix proteins, such as interstitial collagen types I, III, IV, and laminin, and are responsible for the deposition and accumulation of the majority of the excess extracellular matrix in the fibrotic liver.10 In most cases, fibrosis consists of broad, collagenous fibrous bands surrounding normal hepatic lobules. The increase of the periportal connective tissue may compress portal vein radicles, leading to hypertension in the portal system. Cirrhosis is a common end stage for a number of monogenic disorders that result in hepatic insult owing, in part, to fibrotic and cystic changes.9
Renal cysts can occur as manifestations of both non-genetic and genetic disorders.11,12 In the former case, non-hereditary lesions, consisting of an enlarged kidney filled with large cystic structures, can occur in response to ureteral atresia or obstruction. “Renal dysplasia” is the usual term to describe the abnormal differentiation of renal parenchyma, with the occurrence of microscopic abnormalities such as cystic dilatations and primitive ducts surrounded by connective tissue.13 In some conditions, renal dysplasia has been attributed to abnormal embryonic differentiation or developmental arrest leading to the persistence of mesonephric tissue and primitive or fetal structures. Epithelial cell proliferation is thought to be a prerequisite for cyst expansion,14 with some cysts retaining some of the functional properties of the tubular epithelium, such as active ion transport, from which they were derived.15 Cysts that lose their connection with their tubule of origin are thought to expand by a mechanism of transepithelial fluid secretion,16 which cause compression and eventual atrophy of surrounding parenchyma. It is not clear if only a proportion of nephrons contribute to cyst formation, or if all nephrons are involved with varying degrees of dilatation.
FIBROCYSTIC DISEASES OF VISCERAL ORGANS
Polycystic kidney disease in children
“Polycystic disease” was first described as long ago as 1856,17 and a spectrum of disease is now known to exist. Polycystic kidney disease includes autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD). Congenital hepatic fibrosis is a characteristic feature of ARPKD, but may also be associated with other conditions. Liver histology shows portal spaces enlarged by periportal fibrosis, proliferation of biliary ducts, which can be dilated and tortuous, and ductal plate malformation or insufficient remodelling of the primitive intrahepatic biliary system. In some cases, abnormal multiple bile ductules can lose connection with the biliary system and dilate to form large cysts.7 Patients with ARPKD tend to present with congenital hepatic fibrosis and Caroli disease (known as the “combined” form of the disease or Caroli syndrome). The “pure” form of Caroli disease is characterised by multifocal dilatations of the larger, segmental intrahepatic bile ducts. Subsequently, patients can present with recurrent bacterial cholangitis because of bile stagnation at the ectopias of the intrahepatic biliary tree.
The main clinical manifestation of ARPKD is both ectasia and cystic dilatation of the renal collecting tubules in the kidney that prevents the normal development of nephrons, so that they remain as primitive ducts lined by undifferentiated epithelium and sheathed with thick layers of connective tissue. The kidney appears spongy, and there is no clear separation of the cortex and medulla (fig 2D). The prognosis of ARPKD is dependent on the degree of renal involvement and speed of disease progression to renal insufficiency and end stage renal failure (ESRF).18 A number of long term survivors have been reported.
The PCK rat model of polycystic kidney disease also develops renal and hepatic cysts, but in addition also displays ductal plate malformation of the intrahepatic bile ducts and overgrowth of portal connective tissue that resembles human ARPKD.19,20 Genetic analysis of the PCK rat has recently led to the identification of the human orthologue, PKHD1, of the rat gene.21 The human PKHD1 gene is predicted to encode a large protein of approximately 450 kDa, named fibrocystin, of unknown function (fig 3). Fibrocystin contains at least 10 copies of an immunoglobin fold-like domain (the IPT/TIG domain) found in plexins, transcription factors, and extracellular regions of receptor proteins that appear to regulate cell proliferation and cellular adhesion. Another group reported recent similar findings,22 with the identification of a PKHD1 gene product essentially identical, in both size and sequence, to fibrocystin that they name “polyductin”. It will be interesting to elucidate the normal role of fibrocystin in the development and differentiation of collecting ducts in the kidney and bile ducts in the liver. Fibrocystin may be a receptor of growth signals because of the predicted extracellular domain, but could also participate in mediating cell adhesion. The unusual multiplicity of splicing variants for PKHD121,22 may be an important mechanism in regulating these functions by different protein isoforms.

{kind=link}
{kind=link}
{kind=link}
Schematic diagrams of the protein domain structures of polycystins, fibrocystin, and nephrocystin. Coloured boxes or circles on the main backbone of the proteins (thick black line) indicate conserved domains or regions that have a high degree of homology with known proteins. Annotation of domains and regions is based on primary published reports24,263031 and conceptual translations of entries in the SwissProt and Genbank databases for the following accession numbers: NP_000287 (polycystin-1), Q13563 (polycystin-2), O15259 (nephrocystin), and AAL74290 (fibrocystin). The boundaries of selected regions are given as the number of amino acid residues from the N-terminus of the protein (N). The approximate location of other protein domains and putative N-glycosylation sites are based on output from the CDART tool at <www.ncbi.nlm.nih.gov/Structure/lexington/>. Putative transmembrane domains are indicated by grey rectangles, spanning the cell membrane (wavy horizontal line). Regions that have no apparent domains or conserved regions have been left blank.
In contrast, patients with ADPKD tend to present with large hepatic cysts (fig 2C), and the number of hepatic cysts can increase with the age of the patient, but the biliary malformations and hepatic fibrosis that are characteristic of ARPKD are absent in the majority of cases. Defects in two genes have been implicated in ADPKD. Mutations in PKD1, localised at chromosome 16p13,23 appear to cause a more frequent and severe form of ADPKD in comparison to PKD2 at chromosome 4q21–q23. PKD1 encodes an integral membrane glycoprotein, polycystin-1, that is implicated in cell-cell or cell-matrix interactions (fig 3). Polycystin-1 consists of multiple transmembrane domains and a N-terminal extracellular region that probably binds ligands in the extracellular compartment. The C-terminal cytoplasmic region of polycystin-1 contains phosphorylation sites and consensus sequences for a number of signalling molecules, hence suggesting a role in intracellular signal transduction for this protein. The PKD2 gene product, polycystin-2, has significant homology to a voltage-activated Ca2+ channel in the intracellular C-terminal domain (fig 3). Polycystin-1 and -2 appear to interact to form a heterodimeric ion channel at the plasma membrane that regulates renal tubular morphology and function.24
Other renal cystic diseases in children
Medullary cystic kidney disease (MCKD) and juvenile nephronophthisis (NPHP) have different modes of inheritance and different age of onset, but otherwise resemble one another. Corticomedullary cysts, fibrosis, and progressive renal failure are common features of MCKD. MCKD1 affects mainly adults, has an autosomal dominant inheritance pattern, and maps to chromosome 1q21.25 The juvenile form, MCKD2, has recently been shown to arise from mutations in the UMOD gene, encoding the glycoprotein uromodulin.26 In contrast, juvenile nephronophthisis is recessive and two genes (NPHP1 and NPHP4) have been identified recently.27–29 Two other loci have been described for autosomal recessive nephronophthisis that affect children of different age groups.30 It is interesting to note that patients with Senior-Løken syndrome (SLS), which is an association of nephronophthisis and retinal dystrophy, were found to have identical mutations in either the NPHP1 or NPHP4 genes to the affected children with purely renal involvement. This variable phenotype may indicate the action of some modifier locus, although the presence of intrafamilial variation suggests that all patients may have some degree of retinal involvement.
Both NPHP and MCKD share characteristic features, such as tubular atrophy, cyst development, and fibrosis of interstitial cells, although the kidneys are usually of normal size. Cyst formation tends to occur close to the papillary tips in MCKD and at the corticomedullary border in NPHP. Nephrocystin, the NPHP1 gene product (fig 3), appears have a role in mediating cell adhesion.31 Functional studies of the murine homologue of nephrocystin indicate that it can interact with Crk associated substrate, p130(Cas), a protein known to participate in integrin mediated signal transduction and organisation of the actin cytoskeleton at sites of cell adhesion.32 Nephrocystin also contains a c-src homology 2 (SH2) domain, that is implicated in signal transduction (fig 3). In addition, nephrocystin has been shown to interact with nephrocystin-4,29 the protein product of the NPHP4 gene.
FIBROCYSTIC CHANGES AND MALFORMATION SYNDROMES
Skeletal malformation syndromes
Renal, hepatic, and pancreatic abnormalities are common features of Jeune asphyxiating thoracic dystrophy (JATD), although the consistent features are skeletal abnormalities, including a long and narrow thorax, metaphyseal irregularities, and shortness of the ribs and long bones.33,34 Polydactyly of both hands and feet is an occasional feature. Most severely affected cases have a fatal outcome in the perinatal period owing to asphyxia arising from a small thorax and hypoplastic lungs.35 Chronic renal failure occurs in the 20% of patients with JATD who survive beyond the neonatal period. Hepatic involvement can be severe, and can manifest as neonatal cholestasis or hepatic fibrosis, which leads to biliary cirrhosis and portal hypertension.33,36 JATD appears to overlap with other chondrodysplasias in a phenotypic spectrum (table 1).
There are particularly striking similarities between JATD and Ellis-van Creveld syndrome (EvC), which is characterised by short limbs, short ribs, postaxial polydactyly, and dysplastic nails and teeth.37 A differential diagnosis of JATD or EvC, on the basis of radiology alone, may not be possible.38 However, polydactyly, fingernail dysplasia, and congenital cardiac defects, usually a defect of primary atrial septation that forms a common atrium, are the primary features of EvC. In contrast, the main abnormality of JATD is renal. As for JATD, there are occasional developmental defects and dysplasias in the kidneys and liver for EvC.39 It has been suggested previously that JATD, EvC, and hepatic-pancreato-renal (HPRS) syndrome have overlapping features from one disease spectrum.2 The EVC gene has been mapped recently to chromosome 4p1640 and the EVC protein has distant homology to the serine/threonine protein kinase family of proteins and contains a potential transmembrane region, but has at the present time no known function.
Bardet-Biedl syndrome (BBS) is an autosomal recessive disorder that displays pronounced phenotypic variability, with both developmental and progressive defects. The clinical manifestations are difficult to classify, but are characterised primarily by retinal dystrophy, obesity, and postaxial polydactyly.41,42 Renal structural abnormalities, such as multicystic dysplasia, are a minor clinical feature of BBS and may not give rise to clinical evidence of renal disease. At least seven BBS loci have been identified, and the BBS6, BBS2, and BBS4 genes have been identified.41 The BBS4 gene product has significant homology to O linked N-acetylglucosamine transferases, and the BBS4 protein may glycosylate proteins to modulate signal transduction during normal development.
Central nervous system malformation syndromes
Johann F Meckel first described an unusual association of renal cysts, polydactyly, and posterior encephalocele in 1822.43 Subsequently, this association was studied further by Georg B Gruber who used the label “dysencephalia splanchnocystica”.44 Meckel-Gruber syndrome (MKS) is a lethal malformation syndrome characterised by a frequent triad of central nervous system malformations (prosencephalic dysgenesis, occipital encephalocele, and rhombic roof dysgenesis), large multicystic kidneys, bilateral postaxial polydactyly, and fibrocystic changes of the liver45,46 (fig 2B, E). The rates of both cell proliferation and apoptosis were high in the remodelling ductal plate, and moderate in remodelled bile ducts.47 However, the malformed ductal plates in fetal livers with MKS had a low rate of apoptosis, and a lower expression of Fas, a transmembrane receptor that mediates apoptotic signals. In addition, MKS ductal plates had a dramatic increase in proliferative activity and high expression of Bcl-2, an anti-apoptotic protein, in comparison to control livers.47 It is particularly interesting that MKS, JATD, and HPRS syndromes are associated with postaxial polydactyly.48 This common malformation is suggestive that an imbalance between proliferation and cell death exists in these HRFC syndromes, since this could induce the apical ridge to produce an additional digit during limb morphogenesis. There appears to be considerable clinical and genetic heterogeneity in MKS,46,49 with at least three loci linked to the typical MKS phenotype of CNS malformations, cystic kidneys, and polydactyly,50–52 but none of the MKS genes have yet been identified. An alternative, although uncommon, neuropathological anomaly in MKS is a posterior fossa cyst in the form of a cerebellar Dandy-Walker malformation.53 Both the Dandy-Walker malformation and survival beyond the perinatal period are unusual findings in MKS, and may be characteristic of a distinct entity, such as cerebra-reno-digital or Goldston syndromes”.54
The Dandy-Walker malformation, in association with aplasia or severe hypoplasia of the cerebellar vermis, is a feature of other conditions such as HPRS (table 1),56 short rib-polydactyly syndrome type II,57 and Joubert syndrome (JS).58,59 In JS, which is characterised by cerebellar hypoplasia, retinal dystrophy, and impaired psychomotor development, 35% of the subset of JS patients with retinal dystrophy develop renal cysts. The developmental link between the kidney and the CNS may depend upon inductive intercellular interactions during organogenesis60 and a complex interplay between the c-ret proto-oncogene product, fibroblast growth factor receptor (FGF-R), and glial derived nerve factor (GDNF). All of these proteins are expressed in the embryonic renal and CNS tissues and appear to be important during organogenesis.61,62
FIBROCYSTIC CHANGES AND METABOLIC CONDITIONS
The association of renal cystic changes and liver disease is also a feature of a number of inherited metabolic disorders (table 2), and it is therefore important to differentiate these syndromes, which can be readily tested by biochemical means, from recessive HRFC syndromes. For example, Smith-Lemli-Opitz syndrome (SLOS) is caused by the deficiency of 7-dehydrocholesterol reductase (DHCR7), and the clinical features of SLOS include structural brain anomalies such as holoprosencephaly, microcephaly, cleft palate, syndactyly and postaxial polydactyly, cholestatic liver disease, and renal multicystic disease. Renal disease is characterised by microcysts or tubular dilatations. It is particularly interesting that the pattern of anomalies seen for Smith-Lemli-Opitz syndrome (SLOS), such as cerebellar vermis aplasia and renal cystic changes, are similar to those for MKS.63,64 SLOS formes frustes subjects appear to have an even closer overlap with MKS. The discovery of the biochemical defect in this condition has led to the improved management of affected patients and emphasised the importance of sterols in embryogenesis, namely that cholesterol has a direct involvement in the hedgehog embryonic signalling pathway.65
Summary of hepatic and renal anomalies associated with inherited metabolic disorders
Glutaric acidaemia type II (GA2 or multiple acyl CoA dehydrogenase deficiency) is a mitochondrial disorder that results from a deficiency of either electron transfer flavoprotein (ETF) or ETF-ubiquinone oxidoreductase (ETF-QO). Both are intermediate electron carriers to ubiquinone in the mitochondrial respiratory chain. The complete deficiency of ETF-QO is associated with multiple congenital anomalies, including cystic dysplasia of the kidneys, which has led to the misdiagnosis of MKS in the past. CNS anomalies include dysplasia of the cerebral cortex and abnormal neuronal migration, and liver histology typically shows microvesicular steatosis and fibrosis.66 Bile duct hypoplasia and cholestasis may also occur.
Zellweger syndrome (ZS), or cerebrohepatorenal syndrome, is the most severe variant of the Zellweger spectrum of disorders.67 Patients have severe neurological deficit, progressive hepatic and renal dysfunction, and skeletal abnormalities. Renal cysts can vary from glomerular microcysts to large cortical cysts of glomerular or tubular origin. The liver includes architectural abnormalities that disrupt the arrangement and spacing of portal and central areas, with multiple central veins in each hepatic lobule. There may be a paucity of the intrahepatic bile ducts, and either severe fibrosis or micronodular cirrhosis in more advanced cases.
ANIMAL MODELS OF FIBROCYSTIC SYNDROMES
A number of animal models of fibrocystic diseases have been developed, including models of polycystic diseases that involve the liver and biliary tree. Mice with an insertional mutation68 or gene targeting69 of the Pkd1 gene have a phenotype similar to human autosomal recessive polycystic kidney disease (ARPKD). The cpk (congenital polycystic kidneys) mutation on chromosome 12 in mice produces a lethal recessive form of PKD that is associated with hepatic cysts, and has been used as an animal model of human polycystic disease.70 The mouse cpk mutation is also associated with biliary ductal plate malformations, although the expression of the defect was modulated by the genetic background. A second, spontaneous recessive polycystic kidney mutation is jck (juvenile cystic kidneys),71 and a third, the bpk mutation in BALB/c mice, associates with renal cysts and epithelial hyperplasia of the intra- and extrahepatic biliary tracts.72
Transgenic mouse embryos that overexpress keratinocyte growth factor (KGF) develop hepatomegaly, biliary hyperplasia, and cystic dilatation of renal collecting tubules resulting in polycystic kidneys.73 The correct expression of KGF is thought to be important for the mesenchymal-epithelial signalling required during normal embryogenesis.73 However, transgenic mice that overexpress the c-myc proto-oncogene, a regulator of cell proliferation, develop polycystic kidneys,74 as do knock out mice homozygous for deletions of the bcl-2 gene, which normally regulates apoptosis.75 In the latter two model systems, disruption of apoptosis or proliferation would be a reasonable primary cause of cystogenesis. However, the renal cystic abnormalities could equally arise as a pleiotropic effect of the genetic lesion in the mouse model. Until the molecular mechanisms that regulate epithelial differentiation and the balance of cell proliferation or cell death are known in more detail, it is difficult to assess the importance of the phenotypes in these models.
DISCUSSION
In this review, we have compared the variable pathological processes and phenotypes that are associated with several recessive HRFC syndromes. Several reviews have postulated a spectrum of “fibrocystic syndromes” that have a more or less similar aetiology,1–5 but that then subsequently diverge in terms of disease progression and clinical outcome, with variable presentations of fibrosis, hepatocyte damage, cirrhosis, nephritis, and end stage organ failure. The suggestion has been made that the initial phenotypic similarity of HRFC syndromes, at least in terms of histopathology, may arise from similar causative defects in the embryological pathways of liver and kidney development that define tubular structures in epithelial cells.1,2,4 Developmental defects could therefore involve the disruption of cell-cell or cell-matrix interactions of epithelium and mesenchyme. In particular, biliary dysgenesis appears to be a key manifestation that distinguishes the HRFC syndromes (table 1) from the fibrocystic changes of metabolic conditions. Although renal cystic dysplasia is a common (but not obligate) feature of many conditions (table 1), the co-occurrence of biliary dysgenesis, in a form suggestive of ARPKD, appears to be limited to the group of HRFC syndromes that are inherited in an autosomal recessive pattern. In contrast, biliary dysgenesis is not a feature of metabolic conditions, but congenital hepatic fibrosis, cirrhosis, and renal cystic dysplasia are common end points in the progression of metabolic conditions (table 2). This probably indicates that the latter features are the result of any form of insult to the liver and kidney, whether it is congenital or acquired. Renal cyst formation, for example, could be induced by an energy deficit in the tubular epithelial cells, perhaps by disturbing the balance of cell proliferation and apoptosis, rather than by a specific defect in a developmental pathway. Clinical manifestations in Smith-Lemli-Opitz syndrome (table 2) could be a result of cholesterol deficiency during fetal development.
Three plausible molecular mechanisms could explain the common features of HRFC syndromes: (1) the diverse proteins participate in a default developmental pathway for tubulo-epithelial differentiation during embryogenesis of different organ systems, and that this process can therefore be perturbed by a number of genetic lesions; (2) different proteins interact directly with each other to mediate either adhesion or signal transduction events, or to form a multi-subunit complex; (3) different proteins participate in distinct but complementary pathways. These hypotheses are now testable following the recent identification of the PKHD1, PKD1, PKD2, NPHP1, and NPHP4 genes. The encoded proteins (fibrocystin, polycystin-1 and polycystin-2, nephrocystin and nephrocystin-4, fig 3) appear to have pivotal functional roles in renal epithelial differentiation and organisation by mediating interactions between epithelial cells and interstitial components.
Defects in direct protein-protein interactions are now thought to be the primary cause of the cystic changes of the various types of polycystic kidney diseases and juvenile nephronophthisis (NPHP), but the precise mechanisms remain unclear at present. The loss of contact points and signalling events between cells and either the extracellular matrix (at focal adhesions) or other cells (at adherens junctions) may be the defect that underlies NPHP. Nephrocystin, the product of the nephronophthisis type I gene (NPHP1), appears to have a fundamental role in mediating cell adhesion.31 In addition, the C-terminal region of polycystin-1 has been shown to form multi-subunit complexes with focal adhesion proteins that include the structural and actin binding proteins vinculin, talin, tensin, and α-actinin, the adaptor proteins paxillin and p130(Cas), and the signalling kinase c-src.76,77 Polycystin-1 is also thought to form a heterodimeric ion channel with polycystin-2 (see above). The complex of focal adhesion proteins links the extracellular matrix to the actin cytoskeleton through the cell membrane. It is possible that mutations in other proteins of the polycystin-1 complex may lead to either cystic kidney disease or additional extrarenal fibrocystic changes. Familial focal segmental glomerulosclerosis, for example, is thought to be caused by mutations in the focal adhesion protein, α-actinin-4.78 The very recent demonstration, by a series of immunoprecipitation experiments, that nephrocystin and nephrocystin-4 interact directly29 suggests that these proteins are either in the same multi-subunit complex, or interact in a developmental pathway to mediate either adhesion or signal transduction events. Defects in either of the proteins will therefore result in NPHP. A similar molecular explanation has been suggested for the existence of at least six loci or genes for Bardet-Biedl syndrome.41BBS6 encodes a protein that has significant sequence homologies to a class of chaperonins that facilitate protein folding, and other BBS proteins could either form part of a chaperonin complex or interact with BBS6 while still unfolded polypeptides. It will be interesting to determine the causes of genetic heterogeneity in Meckel-Gruber syndrome and Joubert syndrome: do the different encoded proteins interact or participate in the same developmental pathways?
In the future, further study of the structure and function of known proteins, such as fibrocystin and nephrocystin, will contribute to our knowledge of renal cystic dysplasia and biliary dysgenesis. It will then be clear whether or not a common pathogenetic pathway is involved in the range of congenital fibrocystic syndromes, and perhaps provide molecular mechanisms to explain the variability in the rates of disease progression and the severity of clinical phenotype. Understanding these mechanisms may allow novel pharmacological interventions or gene therapy procedures to be designed that would prevent renal cyst development, dysgenesis of the intrahepatic biliary tree, and hepatic fibrosis.
CONCLUSION
This review has identified a number of hepatorenal fibrocystic (HRFC) syndromes that are a group of severe malformation syndromes that cause fibrocystic changes in the liver and kidney, and that are inherited in an autosomal recessive manner. Biliary dysgenesis is a common feature of these conditions, which appears to differentiate them from metabolic conditions with fibrocystic changes. This diversity emphasises the common nature of the embryogenesis in different organ systems, and suggests that tubulo-epithelial differentiation in the kidney and intrahepatic biliary system has a common molecular mechanism. The possible functions of the gene products in these pathways may involve signal transduction by both the extracellular matrix and soluble growth factors, and regulation of epithelial cell proliferation and complex epithelial-mesenchymal interactions that are disrupted in HRFC syndromes. The identification of these genes, and the assessment of the role of mutations, will be an important step in describing critical developmental pathways in molecular detail. Recent work has identified a number of integral membrane proteins that appear to participate in these pathways, but their exact molecular functions remain to be elucidated.
Acknowledgments
The authors wish to thank Professor Deirdre A Kelly and Dr David Milford for helpful discussion and for critical reading of the manuscript. CAJ receives financial support from the Birmingham Children’s Hospital Endowment Fund and the Birth Defects Foundation (grant 02/02). PG is a Royal College of Paediatrics and Child Health/Children Nationwide clinical research fellow in paediatric hepatology.