Article Text

Abstract
Objective To investigate the utility of autozygome analysis and exome sequencing in a cohort of patients with suspected or confirmed mitochondrial encephalomyopathy.
Methods Autozygome was used to highlight candidate genes for direct sequencing in 10 probands, all born to consanguineous parents. Autozygome was also used to filter the variants from exome sequencing of four probands.
Results In addition to revealing mutations in known mitochondrial genes, the analysis revealed the identification of two novel candidate disease genes: MFF and FARS2, encoding the mitochondrial fission factor and phenylalanyl-tRNA synthetase, respectively.
Interpretation These findings expand the repertoire of genes that are mutated in patients with mitochondrial disorders and highlight the value of integrating genomic approaches in the evaluation of these patients.
- Fission
- aminoacyl-transfer RNA
- autozygome
- exome sequencing
- genetics
Statistics from Altmetric.com
Introduction
Mitochondria are unique organelles that typify symbiosis at the cellular level.1 They provide energy to the cell, which in turn provides mitochondria with the bulk of the machinery required for their metabolic processes, including energy production.2 In each mitochondrion, the inner membrane (mitochondria are double-membrane organelles) hosts a complex of proteins that perform a series of redox reactions known as the electron transport chain (ETC) through which energy is extracted from the tricarboxylic acid cycle (TCA)-generated NADH and succinate in the form of ATP and H2O. Each of the five respiratory chain complexes that comprise this chain is composed of a number of proteins, most of which are encoded by nuclear genes.3 Another clinically relevant aspect of the symbiotic relationship between the cell and its dweller mitochondria is the contribution of nuclear genes to mitochondrial translation. Although mitochondria conduct their own translation (they also have their own replication and transcription) with mitochondria-encoded tRNA and rRNA, the nuclear genes encode the factors required for each of the steps of translation (initiation, elongation, and termination), including the synthesis of aminoacyl-tRNA synthetases that serve as enzymatic catalysts in the attachment of amino acids to their cognate mitochondrial tRNA.3 4
This complex interaction between mitochondria and the cell provides the basis for understanding the pathogenesis of mitochondrial disorders in humans.3 Although many cellular proteins are localised in the mitochondria (solely or as part of a wider profile of cellular localisation), mitochondrial disorders is a designation conventionally used when the phenotype is caused by a primary mitochondrial defect, usually reflected in abnormal energy metabolism.3 5 The distribution of tissues that are affected is very wide and is generally believed to mirror the energy demand and, in the case of mitochondrial DNA defects, tissue-specific mutational load. Nonetheless, encephalomyopathy is one of the most common clinical presentations of mitochondrial disorders in paediatrics and is usually, but not necessarily, accompanied by lactic acidosis.6
The clinical workup of patients with suspected mitochondrial disorders is usually extensive and is aimed at assessing the extent of multi-organ involvement as well as specific assays of ETC based on muscle or skin biopsy and sequencing of the 16 kb mitochondrial genome. Ideally, the workup will culminate in the identification of a specific genetic lesion that can inform genetic counselling or even therapy in selected cases.7 However, this ideal outcome is riddled with challenges. For example, despite a classical energy deficiency phenotype, many patients lack conclusive evidence of a specific or global ETC defect on biopsy samples, and while heteroplasmy can be invoked as an explanation in some cases, there must be other explanations when the mutation is in the nuclear DNA. Even when a specific ETC defect is identified—complex I being the most common—the number of genes that encode proteins that are either integral to the complex in question or have an accessory role is prohibitive for conventional clinical sequencing. Moreover, even when a very comprehensive approach is undertaken, where all known and predicted mitochondrial disease genes are sequenced, almost one in two patients remain molecularly undiagnosed—a testimony to the remarkable genetic heterogeneity that characterises these disorders and the need for continued research to uncover additional disease genes.5 6 8
Autozygome analysis is an effective tool in the study of genetically heterogeneous conditions when the parents are related.9 10 Its combined use with exome analysis has proven very powerful in the identification of novel disease genes, even in simplex cases.11 12 In this study, we describe a selected cohort of patients with suspected or proven mitochondrial encephalomyopathy, all born to consanguineous parents. Classical use of autozygome analysis to highlight candidate recessive genes led to the identification of the causative mutation in only a small minority. However, when combined with exome sequencing, it facilitated the identification of the causative mutation in three of the four tested, including two novel candidate disease genes. Our results expand the genetic heterogeneity of these disorders and highlight the utility of combining genomic tools in the study of patients with these disorders.
Methods
Human subjects
We recruited patients with a clinical phenotype consistent with mitochondrial energy disorder based on the combination of developmental delay, basal ganglia involvement on brain MRI, abnormal muscle tone, suggestive muscle biopsy histopathology, and lactic acidosis.13 14 Most patients had muscle and/or fibroblast ETC analysis, but an abnormal result was not required for recruitment. Only patients with consanguineous parents were enrolled to enrich for the possibility of nuclear mutations. The study was approved by the King Faisal Specialist Hospital and Research Center Institutional Review Board, and signed written consent was obtained when recruiting patients and their relatives. Venous blood was collected in both EDTA and Na-heparin tubes from patients and in EDTA tubes from their relatives.
Genotyping and homozygosity scan
DNA samples were processed for genotyping on the Axiom SNP chip platform as per the manufacturer's protocol. Genotypes were analysed for runs of homozygosity (ROH) using autoSNPa (http://dna.leeds.ac.uk/autosnpa). ROH that are >2 Mb and span >107 single nucleotide polymorphisms (SNPs) were used as indicative of autozygosity given the consanguineous nature of the parents.
Candidate gene sequencing
OMIM was queried for the word “mitochondria” and all resulting genes with previously reported mutations in mitochondrial disorders were considered candidate genes (table S1). Only candidate genes that reside within the autozgyome of an affected patient were sequenced as explained before.10 Primers were designed using ExonPrimer webtool (http://ihg.helmholtz-muenchen.de/cgi-bin/primer/ExonPrimerUCSC.pl?db=hg19&acc=uc003yod.3) and their sequences and PCR conditions are available upon request.
Exome sequencing
Exome capture was performed using TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) following the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencer. The reads are mapped against UCSC hg19 (http://genome.ucsc.edu/) by BWA (Burrows-Wheeler Aligner) (http://bio-bwa.sourceforge.net/). The SNPs and Indels are detected by SAMTOOLS (http://samtools.sourceforge.net/).
Mitochondria and peroxisome fission assay
Patient and control skin fibroblasts were plated onto coverslip, and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, glutamine, and penicillin and streptomycin for 48 h. Cells were fixed in 3.6% PFA for 10 min, then permeabilised with 0.1% Triton X-100 (Sigma, St. Louis, MO, USA) followed by blocking in goat serum. To visualise the peroxisomes, fixed fibroblast were incubated with anti-PMP70 primary antibody, followed by rhodamine conjugated secondary antiserum. To examine the mitochondria, fibroblasts were stained with 25 nM of Mito-Tracker Green FM dye (Molecular Probes Life Technologies, Grand Island, NY, USA) for 45 min at 37°, followed by fixation in 3.6% PFA for 10 min. The coverslips were mounted in VectaShield containing DAPI (Vector Labs, Burlingame, CA, USA) and cells were observed under a fluorescent microscope (Nikon Eclipse 90i).
Results
Clinical report
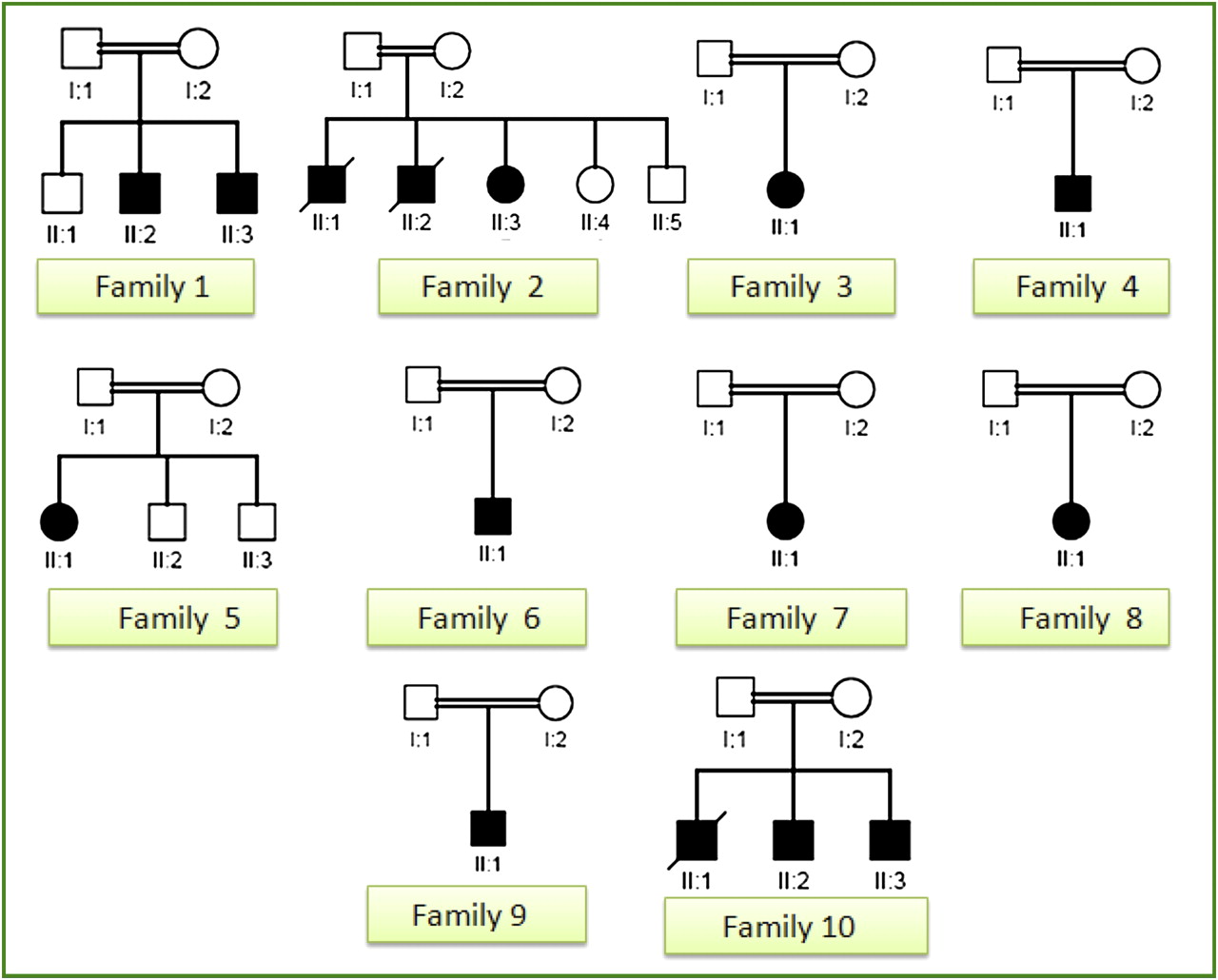
In total, 13 eligible patients were enrolled from 10 families (eight simplex and five multiplex), all born to consanguineous parents (figure 1). Summary of their clinical and laboratory characteristics is provided in table 1 and representative clinical images are shown in figure 2. None of the patients had evidence of point mutation or deletion in their mitochondrial DNA. Detailed clinical notes for the four families in whom a mutation was identified is provided as a supplementary clinical summary.

Pedigrees of 10 probands with mitochondrial encephalomyopathy. Note the consanguineous nature of all study families and the mixed representation of sporadic and multiplex cases.
Clinical, laboratory and pathological characteristics of the study families
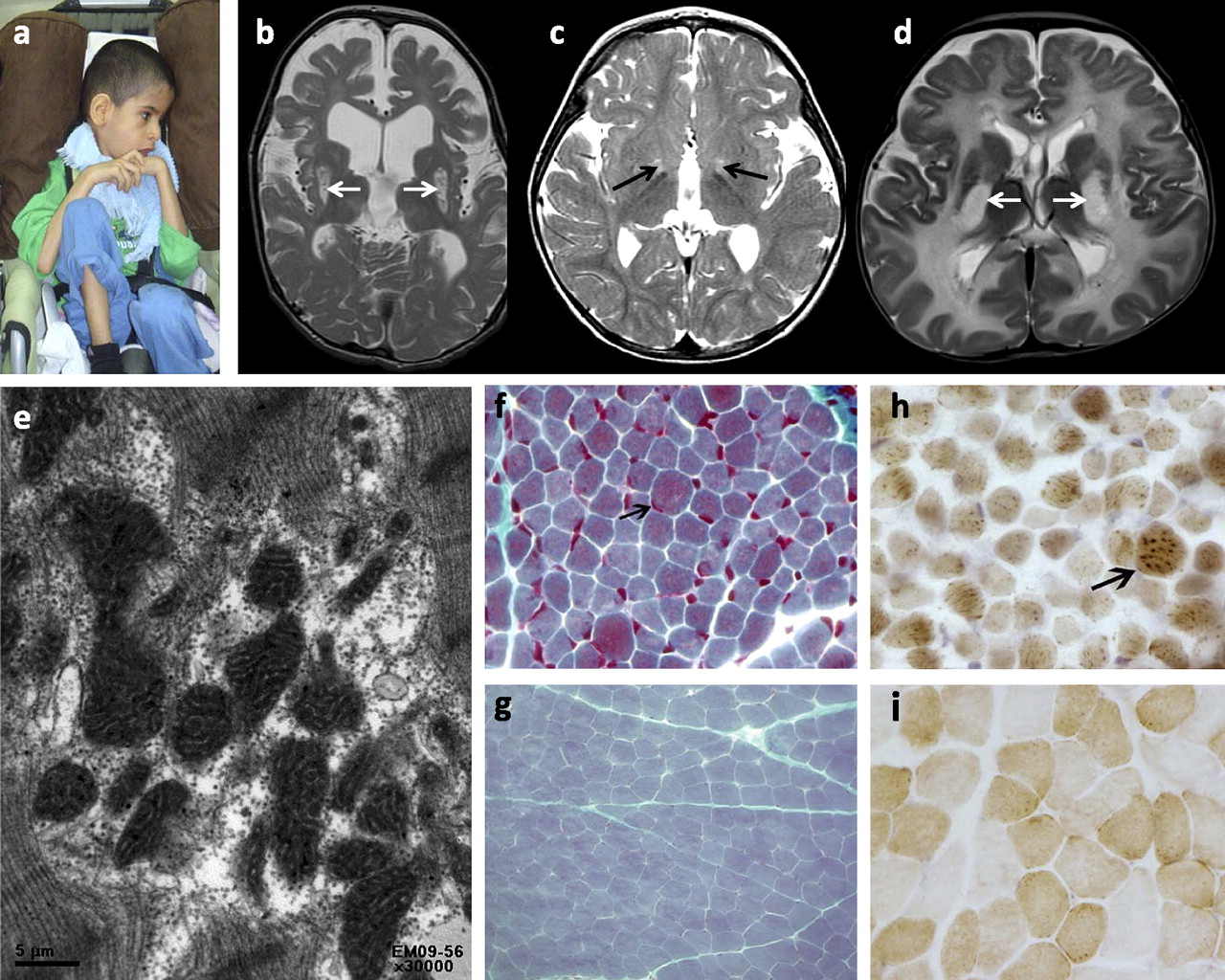
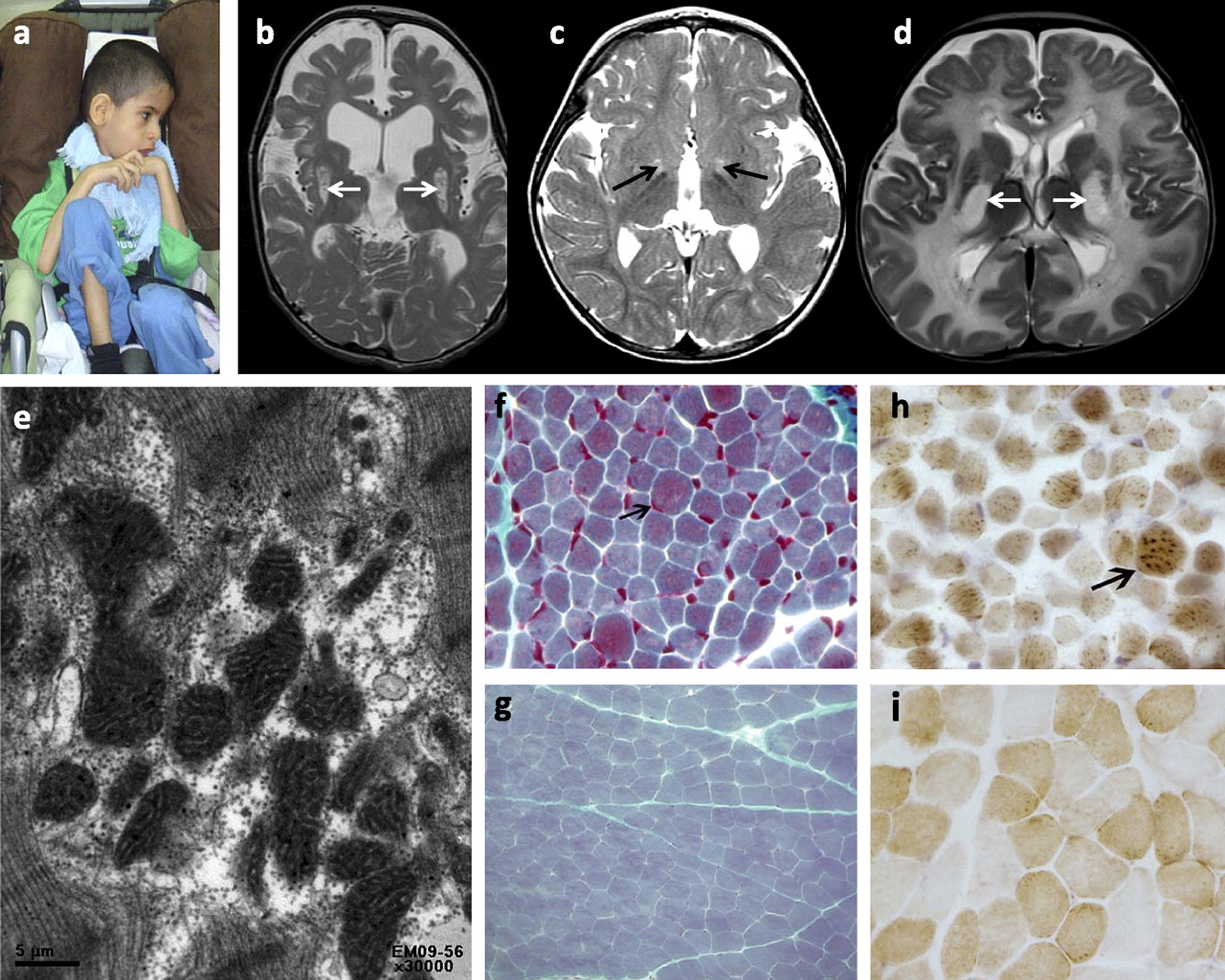
Clinical images of patients with mitochondrial disorders. (A) Proband 3 with emaciation and spastic quadriplegia. (B, C, D) MRI T2 images showing high signal intensity lesions affecting the basal ganglia (arrows) to a varying degree. (E) Electron microscopy image showing irregularly shaped mitochondria with abnormal internal architecture including hypertrophic cristae (×30000). (F) Trichrome stain showing prominent component of red granules (indicating mitochondria) in scattered hypertrophic darkly staining fibres (arrow, ×600) (G is shown to demonstrate the normal staining pattern). (H) Cox stain showing scattered fibres with excessively intense reaction to cytochrome oxidase antibody indicating mitochondrial proliferation and aggregation (arrow, ×600) (I is shown to demonstrate the normal staining pattern).
Autozygome and candidate gene analysis
Consistent with the consanguineous nature of their parents, all subjects had multiple runs of homozygosity (ROH). We considered those that are ≥2 Mb and span >107 SNPs as probably indicative of autozygosity. The entire set of these blocks per individual (autozygome) comprised 7% of his/her genome (range 3–15%). In the first phase of the study, we used the autozygome as a signpost of where a hypothetical autosomal recessive mutation might be so we only sequenced known mitochondrial disease genes that are located within the autozygome of each patient. On average, five genes were sequenced per patient (range 3–7) (figure 3). With the exception of proband 5 in whom we identified a previously reported TSFM mutation (NM_001172696.1: c.997C→T p.R333W), no pathogenic DNA mutation was identified in any of the remaining nine probands.
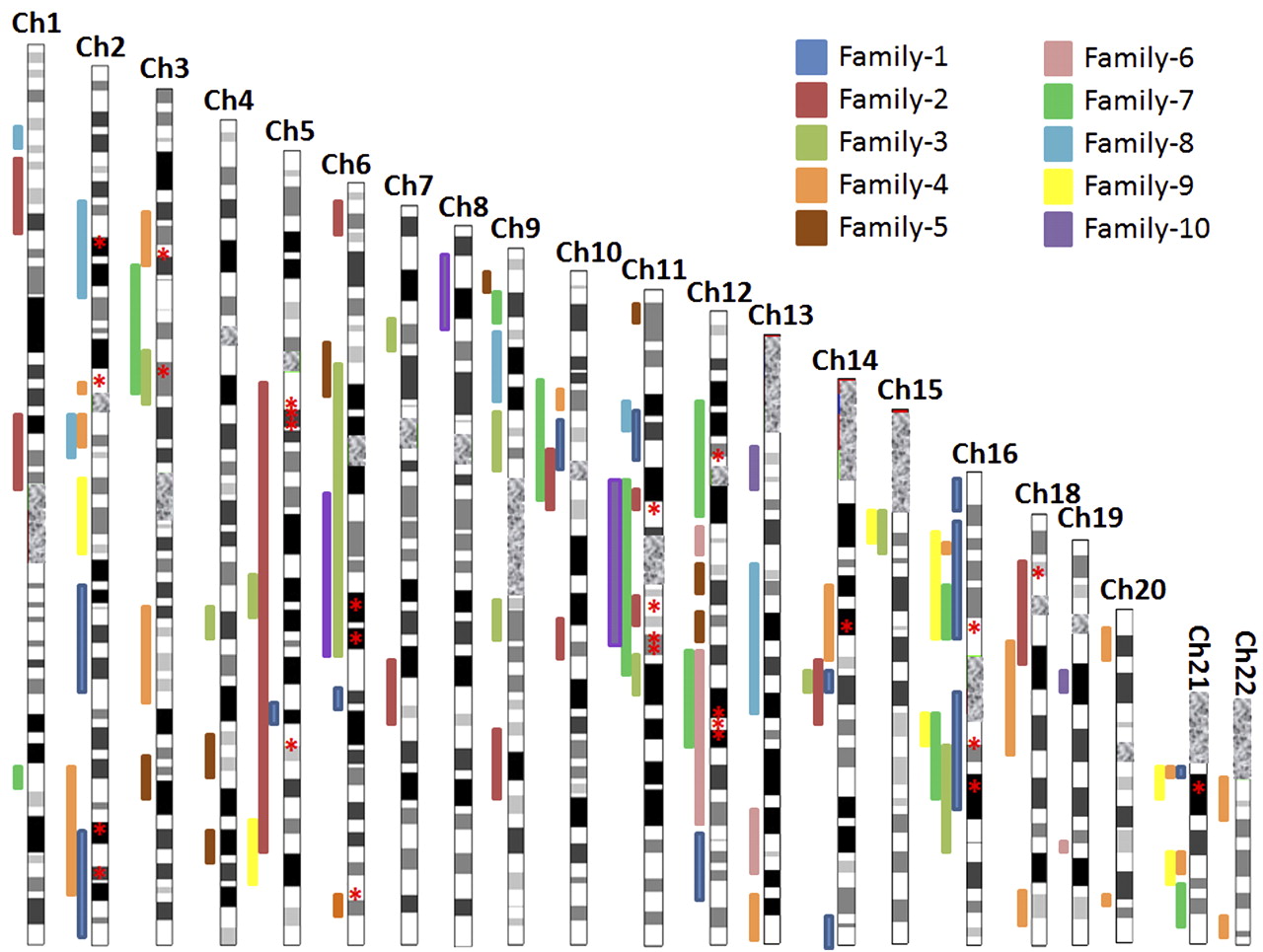
Autozygome analysis of patients with mitochondrial disorders. Bars of autozygosity are superimposed on a chromosome ideogram and are colour coded to match each of the probands. Please note that when more than one sibling is available, we only show bars representative of their shared pattern of autozygosity. Mitochondrial disease genes that have been sequenced are indicated by red asterisks.
Exome analysis
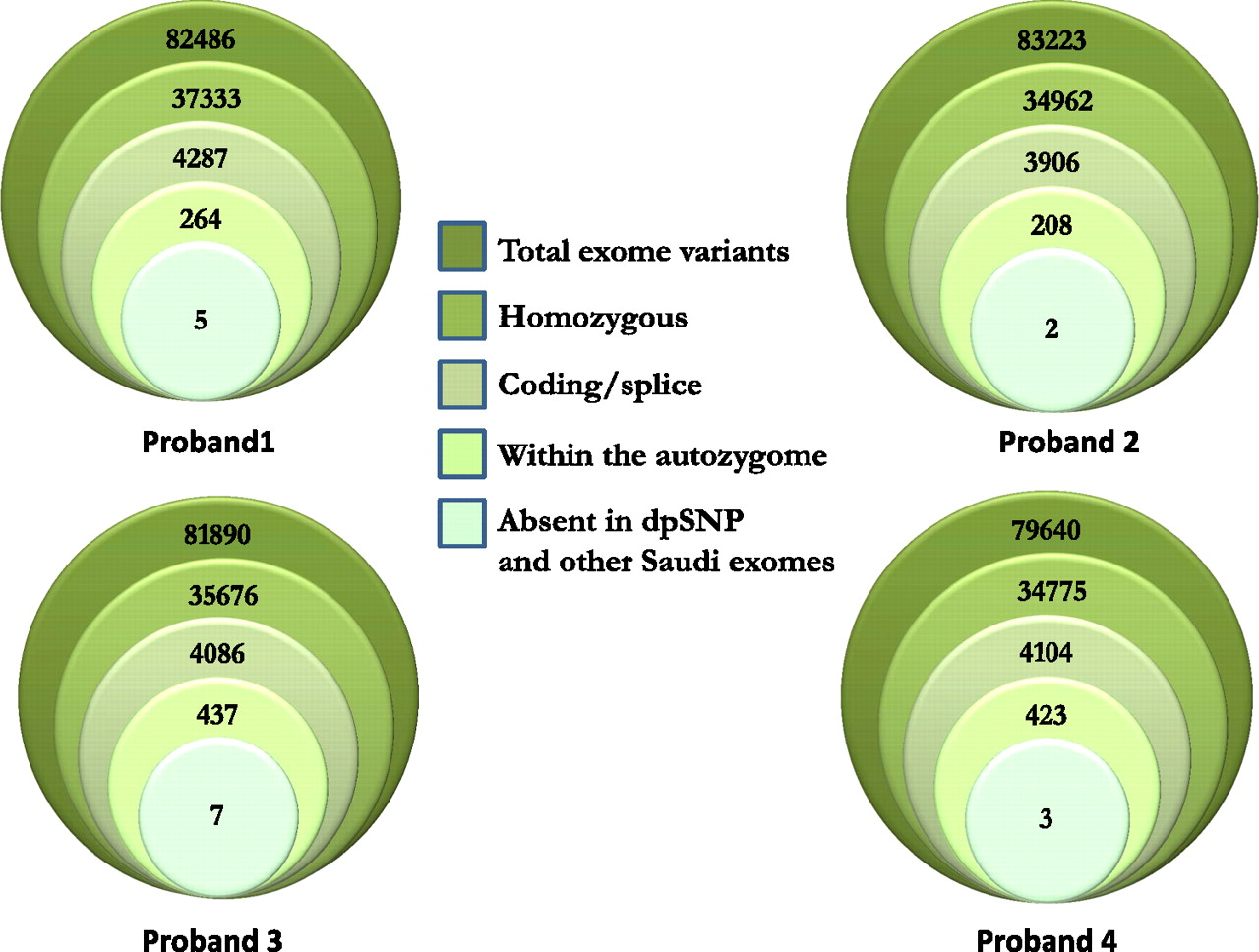
In the second phase of the study, we performed exome sequencing on a randomly selected subgroup comprising four out of the nine probands in whom phase 1 (autozygome/candidate gene) did not reveal the underlying mutation. We only considered novel coding/splicing mutations that are homozygous and that map to the autozygome for each of the four probands (synonymous changes were excluded). Figure 4 and table S2 summarise the resulting variants. Surprisingly, applying this filter revealed the presence of a novel mutation in NDUFS1 (NM_001199981:c.650T→G, p.V217G) in proband 4. This gene was specifically sequenced in phase 1 of this study since this patient had severe complex I deficiency on fibroblast ETC assay, but no pathogenic variants were detected. The mutation that was revealed by exome sequencing involved an exon that was not included in the transcript we used to design sequencing primers, even though both transcripts are RefSeq protein coding transcripts. This highlights a potential pitfall when designing sequencing primers for a large number of candidate genes. This mutation was absent in 114 Saudi controls and is predicted to be pathogenic on PolyPhen.

Exome sequencing and filtration scheme. The total number of exome variants was progressively reduced by applying various filters as explained in the legend. Details about the final variants in the central circles are provided in table S2.
In proband 1, the filtration strategy highlighted a truncating mutation in MFF, encoding mitochondrial fission factor, a compelling candidate given its established role in regulating mitochondrial fission (see below). Sanger sequencing confirmed the mutation, which segregated appropriately with the disease in the family and was absent in 114 Saudi controls. In proband 2, on the other hand, a missense mutation was identified in FARS2, encoding phenylalanyl-tRNA synthetase, another compelling candidate since mutations in other acyl-tRNA synthetases have been documented in patients with mitochondrial disorders. Again, Sanger sequencing confirmed this change, its segregation with the phenotype in the family and its absence in 114 Saudi controls. Finally, only one variant remained after filtration in proband 3, a missense mutation at the first nucleotide of exon 9 (c.1703G→C) of FBXL4. Although this variant does not affect a particularly conserved amino acid residue, it is predicted to affect splicing efficiency. However, reverse transcriptase PCR revealed no change compared to control (data not shown).
MFF truncation leads to dual fission defect
Marked difference in the mitochondrial and peroxisomal appearance of patient fibroblasts that harbour the MFF truncating mutation was observed compared to controls. The typical punctate appearance of mitochondria and peroxisomes, which is a function of their free ends, was largely replaced by a tubular appearance typical of a shift in the balance between fission and fusion in favour of the former (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MFF truncation leads to dual fission phenotype. (A) Control fibroblast cells show normal granular, punctate pattern on mitochondria using mitotracker. (B) Fibroblast cells from patient 1 showing greatly reduced granular pattern, which is largely replaced by a tubular pattern typical of increased fusion and reduced fission. Similar results are seen when peroxisomes are stained with PMP70 antibody (compare the punctate appearance in control cells (C) and tubular configuration in patient cells (D)).
Discussion
The extreme genetic heterogeneity of mitochondrial disorders and the need to identify additional disease genes prompted Calvo et al to develop an assay in which all known and predicted mitochondrial complex I genes are subjected to targeted high throughput sequencing.15 Although this approach did reveal two novel disease genes, NUBPL and FOXRED1, and the underlying mutation in many patients, almost half of the patients remained without a genetic diagnosis.15 Clearly, much work remains to be done to unravel the additional disease genes that eluded this approach.
The use of the autozygome approach has traditionally led to the identification of novel autosomal recessive mitochondrial genes, provided an adequate number of affected family members is available to narrow the area of overlap between the autozygomes of affected individuals to a manageable size for candidate gene selection.16–21 We and others have previously shown how this approach can be very helpful in prioritising known disease genes for direct sequencing.10 22 However, the results of this study show that the utility of this approach appears limited since it led to the identification of the disease gene in only one of 10 probands (10%), a much lower percentage than what we observed for other genetically heterogeneous conditions.23–25 It is likely that the cause of this discrepancy is related to the extreme locus heterogeneity of mitochondrial disorders compared to the other disorders in which we demonstrated a much higher hit rate. Additionally, it is possible that the non-specific presentation of mitochondrial disorders may have led to diagnostic errors and that some of the probands may not actually have a mitochondrial disease, although this is not likely to be a major factor given the high success rate of the second phase of the study (see below). Finally, we acknowledge that the use of a size cut-off carries the inherent risk of missing mutation harbouring ROH that are too small to be detected. However, the fact that parents are first cousins makes this an unlikely explanation at least in the majority of cases since their offspring tend to have larger ROH.9
As a proof of concept, we attempted exome sequencing in four randomly selected probands as a possible diagnostic method in the setting of mitochondrial disorders, particularly after the recent success we have shown in combining exome and autozygome analysis even in simplex cases. Indeed, this phase of the study showed a much higher success rate as it unravelled the likely disease causal mutation in three out of four probands. Interestingly, the NDUFS1 mutation was missed in the first phase of the study because the transcript in which the exome revealed the causative mutation was not considered in the primer design.
The two novel genes highlighted revealed in phase 2 are compelling candidates. Other than being present in ∼7% of the genome in which we focused our analysis (autozygome), their high pathogenicity score on in silico modelling, and their absence from ethnically matched controls, each of these genes is intricately related to mitochondrial biology. In the case of MFF, this gene was identified on a screen for factors that control mitochondrial fission.26 Mitochondria swing between a state of fusion and fission, two opposite states that need to be precisely balanced. Deficiency of fusion capacity deprives mitochondria of the ability to exchange constituents and has been documented as a cause of mitochondrial diseases in patients.27 On the other hand, the role of fission in mitochondrial diseases was appreciated only recently when a mutation in DRP1, which requires MFF for mediating mitochondrial fission, was found to underlie a unique form of mitochondrial cytopathy that is accompanied by defective peroxisomal fission as well.28 The truncating mutation we identified in MFF removes the transmembrane domain which was found experimentally to completely abolish its mitochondrial function and result in a mitochondrial phenotype consistent with lack of fission.29 Indeed, we have shown a dual mitochondrial and peroxisomal fission defect in patient fibroblasts that is identical to the one described in MFF knockdown and in truncation mapping experiments. Lack of overt peroxisomal disturbance phenotype (concentration of very long chain fatty acids (VLCFA) was normal) is in line with what is reported in the patient with DRP1 mutation who, despite having a dual fission defect in both mitochondria and peroxisomes, only displayed minimal elevation of VLCFA. This suggests that fission defects are more consequential to mitochondria as compared with peroxisomes. Given the readily identifiable mitochondrial fission phenotype in patients with MFF and DRP1 mutations, it is tempting to suggest adding this assay to the various mitochondrial assays that are routinely performed on diagnostic biopsy samples taken from patients with mitochondrial encephalomyopathy.
The mutation in FARS2 involves one of the aminoacyl-tRNA synthetase genes, many of which have been implicated in mitochondrial disorders given their critical role in mitochondrial translation.30–32 Reassuringly, the missense mutation we identified affects an absolutely conserved catalytic domain and is predicted to be detrimental to the function of this enzyme.33 Despite the compelling nature of these two candidate genes, we acknowledge that their unequivocal classification as disease genes in mitochondrial disorders requires the identification of additional pathogenic alleles among patients with similar phenotype, which we hope will be helped by our reporting of these two genes.
Exome sequencing has been used recently to identify novel mitochondrial disease genes in simplex cases using mitochondrial databases or selective targeting of mitochondrial genes (MitoExome).31 34 35 We are in agreement with Calvo et al that integrative genomic approaches are needed to untangle the complex nature of mitochondrial disorders and we hope this study shows the power of combining the two genomic tools exome and autozygome in the study of these disorders.36 Given the apparent success of this approach, we plan to apply it on the remaining subjects and on additional subjects in future studies.
Acknowledgments
We thank the patients and their families for their enthusiastic participation. We thank the Genotyping and Sequencing Core Facilities at KFSHRC for their technical help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement table 1
- Data supplement 2 - Online supplement table 2
Footnotes
Funding This study was funded in part by KACST grants 08-MED497-20 and 09-MED491-20 (FSA) and Dubai-Harvard Foundation for Medical Research Collaborative Grant (FSA).
Competing interests None.
Patient consent Obtained.
Ethics approval King Faisal Specialist Hospital and Research Center Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.