Article Text

Abstract
Background Sensenbrenner syndrome is a heterogeneous ciliopathy that is characterised by skeletal and ectodermal anomalies, accompanied by chronic renal failure, heart defects, liver fibrosis and other features.
Objective To identify an additional causative gene in Sensenbrenner syndrome.
Methods Single nucleotide polymorphism array analysis and standard sequencing techniques were applied to identify the causative gene. The effect of the identified mutation on protein translation was determined by western blot analysis. Antibodies against intraflagellar transport (IFT) proteins were used in ciliated fibroblast cell lines to investigate the molecular consequences of the mutation on ciliary transport.
Results Homozygosity mapping and positional candidate gene sequence analysis were performed in two siblings with Sensenbrenner syndrome of a consanguineous Moroccan family. In both siblings, a homozygous mutation in the initiation codon of C14ORF179 was identified. C14ORF179 encodes IFT43, a subunit of the IFT complex A (IFT-A) machinery of primary cilia. Western blots showed that the mutation disturbs translation of IFT43, inducing the initiation of translation of a shorter protein product from a downstream ATG. The IFT-A protein complex is implicated in retrograde ciliary transport along axonemal microtubules. It was shown that in fibroblasts of one of the siblings affected by Sensenbrenner syndrome, disruption of IFT43 disturbs this transport from the ciliary tip to its base. As anterograde transport in the opposite direction apparently remains functional, the IFT complex B proteins accumulate in the ciliary tip. Interestingly, similar results were obtained using fibroblasts from a patient with Sensenbrenner syndrome with mutations in WDR35/IFT121, encoding another IFT-A subunit.
Conclusions The results indicate that Sensenbrenner syndrome is caused by disrupted IFT-A-mediated retrograde ciliary transport.
- Calcium and bone
- clinical genetics
- molecular genetics
- cell biology
- renal medicine
Statistics from Altmetric.com
Introduction
Sensenbrenner syndrome or cranioectodermal dysplasia (MIM 218330) is a rare autosomal-recessive heterogeneous ciliopathy that is primarily characterised by skeletal abnormalities (eg, craniosynostosis, narrow rib cage, short limbs, brachydactyly) and ectodermal defects.1 2 Nephronophthisis leading to progressive renal failure, hepatic fibrosis, heart defects and retinitis pigmentosa have also been described.3–6 Recently, mutations in two genes have been found to cause Sensenbrenner syndrome, IFT122 (encoding intraflagellar transport (IFT) protein 122; MIM 606045) and WDR35 (encoding WD repeat-containing protein 35; MIM 613602).7 8 Both genes encode proteins that are part of the IFT complex A (IFT-A). This multisubunit complex, consisting of at least six proteins, forms the core of a particle that is mobilised by the cytoplasmic dynein/dynactin motor. The IFT-A particles use the microtubule bundles of the ciliary axoneme to carry membrane vesicles and other ‘cargo’ proteins from the tip of a cilium to its base (retrograde IFT).9 10 Both retrograde and anterograde IFT (transport towards the ciliary tip), are required for ciliary assembly, disassembly and homeostasis.9–11
Methods
Single nucleotide polymorphism (SNP) array analysis
Genomic DNA was isolated from peripheral blood samples of two siblings with Sensenbrenner syndrome and their parents. Each DNA sample was genotyped with an Affymetrix 250K NspI array (Affymetrix, Santa Clara, California, USA), which contains 262 000 SNPs. Array experiments were conducted according to protocols from the manufacturer. The 250K SNP genotypes were analysed with Genotyping Console software (Affymetrix). PLINK12 (available online) was used for calculation of regions of homozygosity.
Mutation analysis
Candidate genes were sequenced using patient genomic DNA. Primers for C14ORF179 and TTC8 were designed with the Primer 3 program (freely available online); primer sequences are listed in online supplementary table 3. All coding exons of C14ORF179 and TTC8 were amplified by PCR and analysed forward and reverse with a dye-termination chemistry (BigDye Terminator, version 3 on a 3730 DNA analyzer; Applied Biosystems, Inc., Foster City, CA, USA). PCR conditions are available upon request.
DNA constructs
A full-length cDNA clone (IRAUp969A1256 from ImaGenes) was used as a template to amplify full-length C14ORF179 (NM_052873, isoform 1) with Gateway compatible primers. A consensus kozak sequence ‘gccgccacc’ or a C14ORF179 5′UTR fragment ‘gtttccaggaagtgacgtcaggcggccgcggag’ was included in the forward primers, just before the ‘atg’. PCR products were used to create Gateway-adapted constructs using the Gateway cloning system (Life Technologies, Bleiswijk, The Netherlands). C14ORF179 was then cloned into two vectors, the Gateway-adapted SF-TAP vector (with a C-terminal Strep II-FLAG-tag)13 and p504 (with a C-terminal eYFP tag).14 Wild-type constructs and vectors containing the translation initiation codon mutation were made.
Transfection and western blot analysis
HEK293T cells (human embryonic kidney) were transfected with plasmid DNA using effectene (Qiagen, Venlo, The Netherlands) in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS), according to the manufacturer's instructions. Twenty-four hours after transfection, cells were lysed in ice cold lysis buffer (1× Tris-buffered saline, 0.5% NP-40 with complete protease inhibitor cocktail, Roche, Woerden, The Netherlands). Lysates containing IFT43-eYFP and IFT-Strep II-FLAG were cleared by centrifugation at 4°C for 10 min at 14 000 g. After adding NuPAGE sample buffer with reducing agent (Invitrogen) to the samples, lysates were heated for 10 min at 70°C. Samples were subsequently analysed by SDS-PAGE (using a NuPAGE Novex 4–12% Bis-Tris SDS-PAGE gel) followed by western blotting using α-FLAG (Flag M2 mouse monoclonal antibody, Sigma, 1:1000, Sigma-Aldrich, Zwijndrecht, The Netherlands) and α-GFP (GFP mouse monoclonal antibody, Roche, 1:1000) antibodies. IRDye800 (Rockland, Gilbertsville, PA, USA) goat anti-mouse IgG was used as a secondary antibody (1:10 000). Blots were washed with phosphate-buffered saline (PBS) with 0.1% Tween-20. Fluorescence was detected on a Li-Cor Odyssey 2.1 infrared scanner (Li-Cor, Lincoln, NE, USA).
Immunocytochemistry
Fibroblast cells from patients with Sensenbrenner syndrome (CL10-00031 and CL10-00021) and a control individual (CL10-00010) were stained with antibodies to the following proteins: IFT88 and IFT57 (rabbit polyclonal; both kindly provided by G Pazour; 1:300 and 1:250, respectively); GT335 (mouse monoclonal antibody; kindly provided by C Janke; 1:1500); RPGRIP1L (guinea pig polyclonal; SNC040, 1:500),14 and acetylated α-tubulin (mouse monoclonal, Life Technologies, Bleiswijk, The Netherlands, 1:1000). The following secondary antibodies (all from Life Technologies, Bleiswijk, The Netherlands) were used: anti-guinea pig IgG Alexa Fluor 568 and 488 (1:300), anti-rabbit IgG Alexa Fluor 568 and 488 (1:300) and anti-mouse IgG Alexa Fluor 405 (1:300). Fibroblast cells were seeded (∼1:3) on sterile cover glasses and incubated for 24 h at standard cell culture conditions in DMEM with 20% FCS. Cells were subsequently serum-starved for 48 h with DMEM with 0.2% FCS to stimulate cilia formation. Cells were then briefly washed in PBS and fixed in 2% paraformaldehyde in PBS for 20 min. Cover glasses were subsequently blocked with freshly made 2% bovine serum albumin in PBS, followed by a 1 h incubation with the primary antibody at room temperature. Cells were washed with PBS and then incubated with secondary antibodies for 30 min. Cover glasses with the stained cells were placed upside down on a drop of Vectashield (Vector Laboratories, Burlingame, CA, USA) on a microscopic glass slide. Microscopic analysis was performed on a Zeiss Axio Imager Z1 fluorescence microscope (Zeiss, Sliedrecht, The Netherlands) with an ApoTome slider. Images were processed with AxioVision (Zeiss) and Photoshop CS4 (Adobe Systems, San Jose, CA, USA).
Accession numbers and web resources
Accession numbers of the C14ORF179 gene and a list of the web resources that were used in this study can be found in online supplementary Materials and Methods.
Results
Clinical studies and genotyping
To identify additional genetic defects underlying Sensenbrenner syndrome, we conducted genome-wide homozygosity mapping using Affymetrix 250K arrays in a consanguineous family from Moroccan descent with two siblings with Sensenbrenner syndrome (family P05-1040, figure 1 and supplementary table 1). Homozygosity analysis by PLINK12 showed that the largest and second-largest homozygous regions of the affected siblings from family P05-1040 (patients II:1 and II:2, table 1) are overlapping; this region was 25.2 Mb on chromosome 14 (containing 158 annotated RefSeq genes), with SNP_A-2107558 and SNP_A-2312334 as bordering SNPs (figure 1, table 1 and supplementary table 2). Other homozygous regions had less overlap, indicating that the genetic defect was most likely to be found in the region on chromosome 14 that contains the genes C14ORF179 and TTC8.

Family P05-1040 is affected by Sensenbrenner syndrome. (A) P05-1040 is a consanguineous family of Moroccan descent with two siblings with Sensenbrenner syndrome. Clinical features of patient II:1 (B-F) and patient II:2 (G-K). (B) Absence of typical craniofacial features. (C) Rhizomelic shortening of limbs, narrow thorax. (D) Hypoplastic, cone shaped and widely spaced teeth. (E) Bilateral 2-3-4 toe syndactyly. (F) Brachydactyly, webbing of fingers, short and broad nails. (G) Frontal bossing, telecanthus, micrognathia, sparse and fine hair. (H) Rhizomelic shortening of limbs, narrow thorax. Haemodialysis catheter, Ciminoshunt left arm, gastrostomy. (I) Hypoplastic and widely spaced teeth. (J) Bilateral postaxial polydactyly, bilateral 2-3 toe and 5-6 toe syndactyly, sandal gap between 1-2 toes. (K) Brachydactyly, webbing of fingers, short and broad nails after surgical correction of postaxial polydactyly. Written informed consent from the parents was obtained to publish images from patients II:1 and II:2.
Homozygosity mapping in Sensenbrenner family P05-1040
Given that IFT122 and WDR35, which are both associated with Sensenbrenner syndrome, are part of the IFT-A protein complex, we selected C14ORF179 as our primary candidate, as this gene encodes the human orthologue of another member of the IFT-A complex, IFT43.9 10 In addition, we selected TTC8 (tetratricopepteide repeat domain 8; MIM 608132) as a candidate gene. This gene encodes an IFT regulator and is disrupted in Bardet–Biedl syndrome (MIM 209900).15 16
Mutation detection
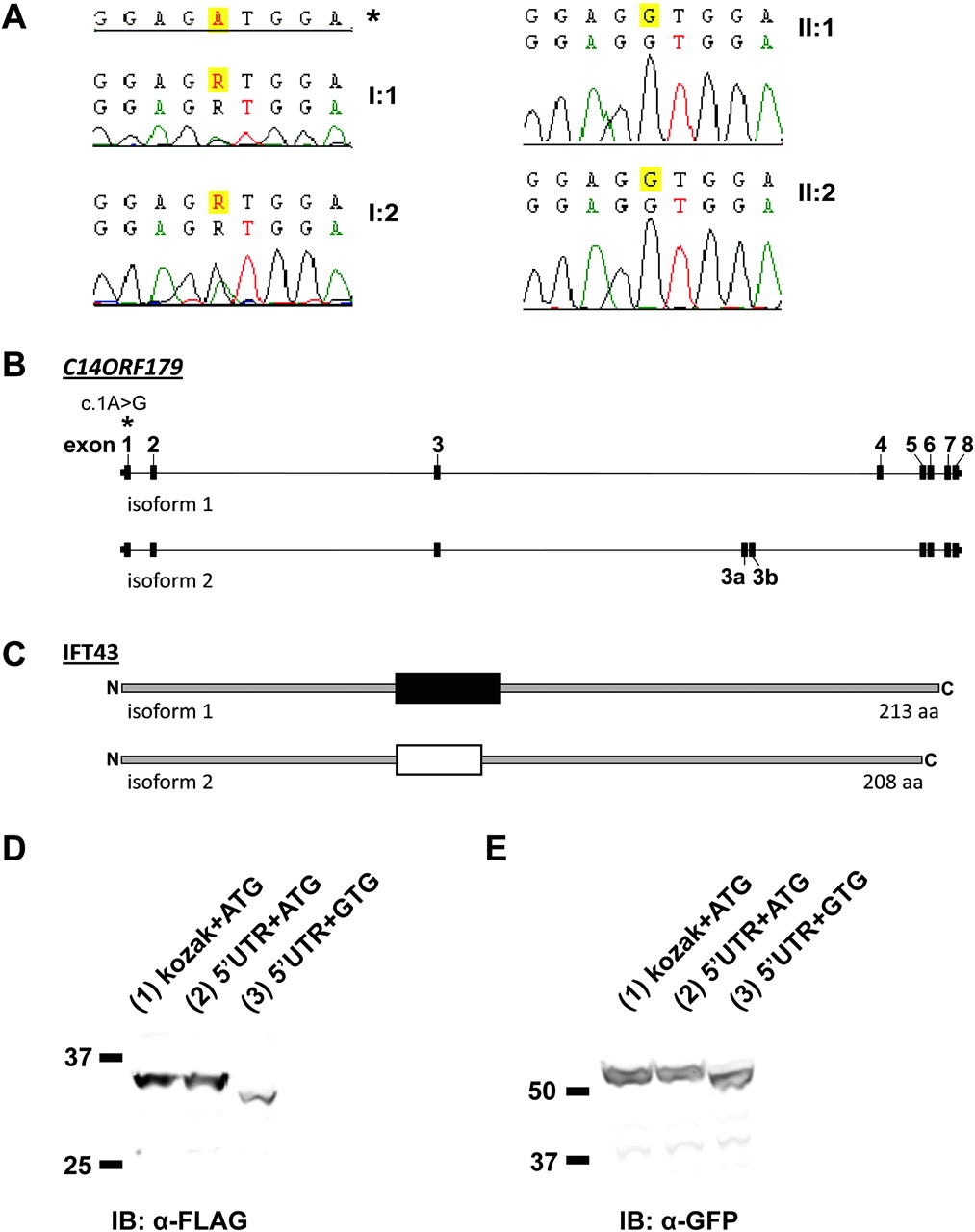
Sequence analysis of candidate genes TTC8 and C14ORF179 in the affected siblings excluded genetic defects in TTC8 as a cause of Sensenbrenner syndrome in these patients, but did show a homozygous variation in the translation initiation codon in exon 1 of C14ORF179 (c.1A→G) (figure 2A). This variation co-segregates with the disorder in the family, as both (related) parents are heterozygous for this variation (figure 2A). The c.1A→G variant was present in neither dbSNP nor in 192 Dutch- and 122 Moroccan control alleles, respectively, which provides further evidence for the pathogenicity of the identified mutation.

Mutations in C14ORF179 cause Sensenbrenner syndrome. (A) In family P05-1040, both affected siblings (II:1 and II:2, right panel) carry a homozygous mutation (c.1A→G) in the translation initiation codon of C14ORF179. Both first cousin parents (I:1 and I:2, left panel) are heterozygous for this variation. The asterisk shows the reference sequence. (B) C14ORF179 contains 10 exons and is alternatively spliced. The mutation (c.1A→G) that was identified in family P05-1040 as a cause of Sensenbrenner syndrome is indicated with an asterisk. (C) Two major splice variants of C14ORF179 encode two different isoforms of IFT43 that vary in their central protein domain. (D, E) Western blot analysis of cell lysates from HEK293 cells expressing recombinant wild-type (lanes 1 and 2) and mutated IFT43 (lane 3), C-terminally fused with a Strep II-FLAG tag (left panel) or an α-GFP tag (right panel). Variations of the initiation codon and upstream sequence of C14ORF179 are indicated. The initiation codon mutation interferes with translation, resulting in a shortened protein (lane 3). The difference between the mutated and the wild-type protein is approximately ∼3 kDa in both α-FLAG and α-GFP immunoblots (compare lanes 2 and 3). A wild-type construct with a kozak sequence (instead of the ′5 UTR fragment of C14ORF179) was used as a control for protein expression.
In an attempt to identify more mutations in C14ORF179, we screened this gene for mutations in four unrelated patients with Sensenbrenner syndrome who did not carry WDR35 mutations. We did not identify mutations in these patients. Since homozygosity mapping in patients from two consanguineous Sensenbrenner families did not reveal major stretches of homozygosity in the regions containing WDR35, IFT122 or C14ORF179, it is likely that the heterogeneity in Sensenbrenner syndrome extends beyond these three genes.
C14ORF179 mutation causes aberrant translation of the encoded IFT43 protein
C14ORF179 contains 10 exons that encode two major protein isoforms derived from alternative splicing that only vary in a central segment of both isoforms (figure 2B,C). Although ATG→GTG mutations have been reported to be pathogenic (eg, in hereditary osteodystrophy,17 Norrie disease18 and β-thalassaemia19), it has also been reported that the GTG codon can be used as an initiation codon, albeit rare and inefficient.20–22 We therefore investigated the effect of the c.1A→G mutation in C14ORF179 on its encoded protein. To test if this mutation diminishes translation or if it leads to translation initiation from a downstream ATG (most likely from the first downstream ATG in exon 2), we cloned the full-length C14ORF179 cDNA with and without the c.1A→G mutation into expression vectors encoding eYFP and Strep II-FLAG fusion tags. Western blot analysis of the recombinant tagged IFT43 proteins shows that the translation initiation codon mutation indeed disturbs translation; introduction of the mutation yields IFT43 proteins with a lower molecular weight of ∼3 kDa compared with wild-type proteins (figure 2D,E). This is in agreement with translation initiation at the second ATG of the coding sequence (in exon 2), at position c.A64, which becomes the first available initiation codon due to the mutation. This results in an N-terminal deletion of 21 amino acids (p.Met1_Val21del) in the same open reading frame.
Mutation in C14ORF179/IFT43 disrupts ciliary transport
The structure of IFT43, the protein product of C14ORF179, remains largely elusive as the protein contains no significant homology with any structural or functional motifs (as determined by SMART23) (figure 2C). The protein was initially studied in Chlamydomonas reinhardtii.9 The Chlamydomonas orthologue is part of the IFT complex A that is involved in retrograde transport in the cilium.9 10 The IFT-A protein complex consists of IFT43, IFT121/WDR35, IFT122, IFT139, IFT140 and IFT144,10 and is associated with THM1 and TULP3.24 Recently, it was shown that IFT43 directly binds to IFT121/WDR35, which supports a tight relationship between these two proteins.25 This is remarkable, since IFT121/WDR35 is the protein product of WDR35, a previously identified Sensenbrenner gene.8
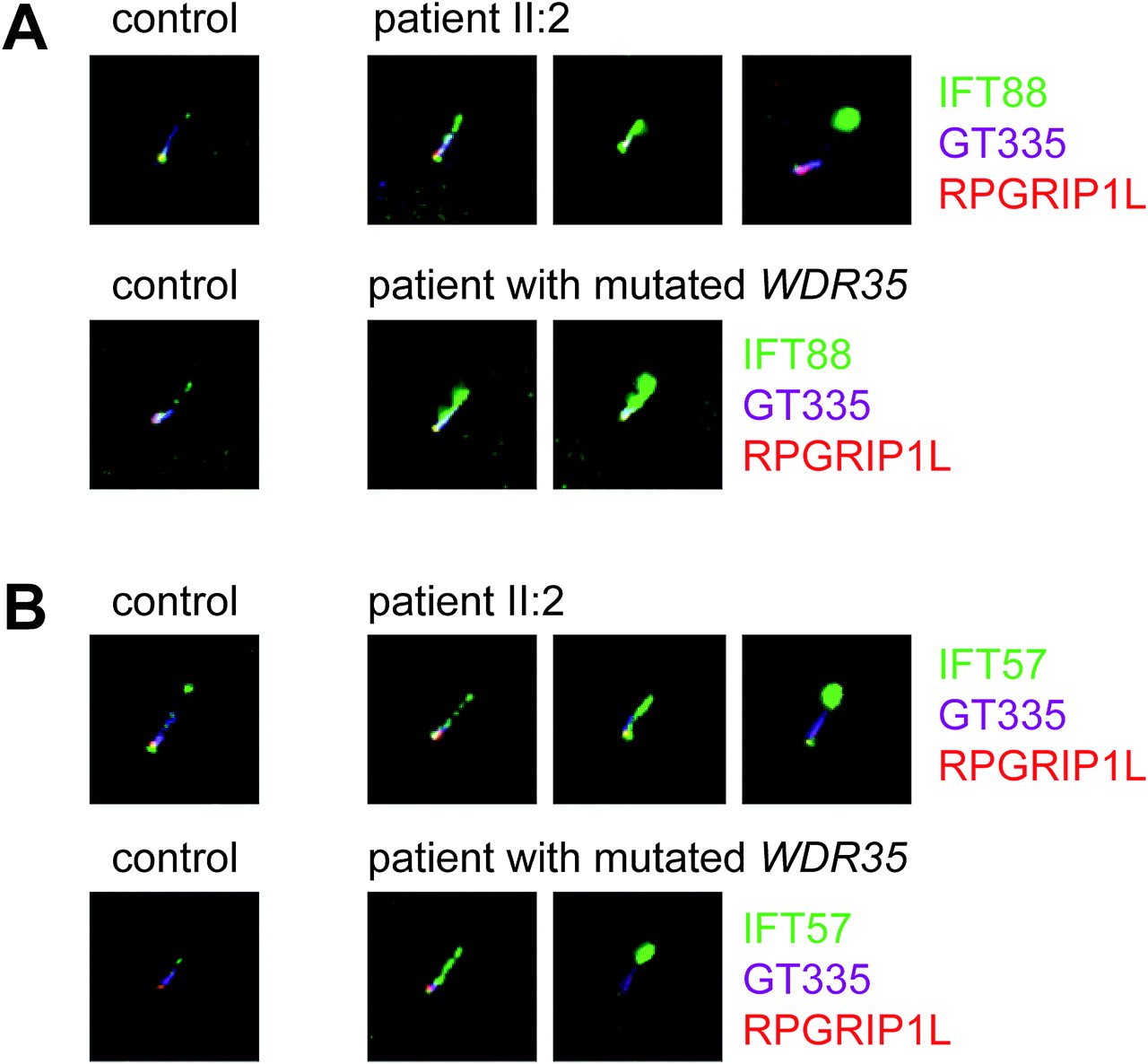
Studies in multiple species, including C reinhardtii, Caenorhabditis elegans, Drosophila melanogaster and Trypanosoma brucei, have shown that disruption of retrograde transport leads to formation of bulged cilia that contain an accumulation of IFT-B complex proteins in their tips.24 26–30 In order to validate the disruptive effect of the identified mutation in C14ORF179 in family P05-1040 (figures 1 and 2), we stained fibroblasts from one of the affected siblings (II:2) using antibodies against the IFT-B complex protein members IFT88 and IFT57. Indeed, we identified a similar accumulation of IFT-B complex proteins in the distal part of the ciliary axoneme and in the ciliary tip (figure 3 and supplementary figures 1 and 2), while in the cilia of control fibroblasts, these proteins were less abundant and primarily localised at the basal body and transition zone.

{kind=link}
{kind=link}
{kind=link}
IFT88 and IFT57 accumulate in distal ends of cilia in fibroblasts from patients with Sensenbrenner syndrome with C14ORF179 and WDR35 mutations. (A) Fibroblasts were stained against the IFT-B complex protein IFT88, GT335 (a marker of the proximal cilium) and RPGRIP1L (a transition zone marker). IFT88 (green); GT335 (purple); and RPGRIP1L (red). Comparison of the IFT88 staining in control fibroblasts with cells from patients with Sensenbrenner syndrome shows that IFT88 accumulates in the ciliary tips of the patient's fibroblasts. Note, that there is a certain extent of variability between IFT defects in cilia from the patients—that is, in some cilia the IFT88 staining is more intense (and more distal) than in others. (B) Fibroblasts from patients with Sensenbrenner syndrome with C14ORF179 and WDR35 mutations were stained for IFT57 (an IFT-B complex protein) and two markers that stain the base of the cilium, GT335 and RPGRIP1L. IFT57 (green); GT335 (purple); and RPGRIP1L (red). Like IFT88, IFT57 accumulates in the distal ends of cilia in fibroblasts from the patients.
Subsequently, we compared these ciliary defects with the phenotype of cilia in fibroblasts from a patient with Sensenbrenner syndrome with WDR35 mutations (a patient who we described previously8) and found nearly identical defects (figure 3 and supplementary figures 1 and 2). Moreover, similar to previous reports on patients with Sensenbrenner syndrome with IFT122 mutations,7 we found that cilia from patient fibroblasts (II:2, figure 3) are somewhat shorter than those of healthy controls (supplementary figure 3).
Discussion
We report on a family with two children with Sensenbrenner syndrome who carry a mutation in the translation initiation codon of C14ORF179. This is the third gene associated with Sensenbrenner syndrome and in line with the other two genes (WDR35 and IFT122), C14ORF179 also encodes a member of the IFT-A particle (IFT43). Mutations in WDR35 can cause both Sensenbrenner syndrome and the more severe (and embryonically lethal) short-rib polydactyly (personal communication, PJ Lockheart). We therefore suggest that both syndromes are part of a phenotypic spectrum of IFT-A complex disruption, similar to previous proposals for Joubert syndrome and allied ciliopathies like Meckel–Grüber syndrome and Bardet–Biedl syndrome. The remaining protein activity then determines the severity of the disease phenotype. Indeed, in all genes associated with Sensenbrenner syndrome identified thus far, only mutations with a relatively mild disruptive character (mainly missense mutations, sometimes in combination with a truncating mutation) have been identified, including the mutation described in this report, which results in a small, N-terminal truncation of IFT43. In this respect, it is interesting to note that although we show that such mild mutations in cilia of patients with Sensenbrenner syndrome fibroblasts disrupt retrograde transport, they do not abrogate ciliogenesis.
Sensenbrenner syndrome clinically overlaps with Jeune syndrome (also known as asphyxiating thoracic dystrophy; MIM 208500). Skeletal anomalies such as brachydactyly, short limbs and a narrow thorax have been reported in both disorders. A skeletal characteristic that is unique for Sensenbrenner syndrome is craniosynostosis; however, only the youngest sibling in family P05-1040 displayed this typical characteristic (supplementary table 1, patient II:2). This complicated the initial clinical diagnosis in this family as it was unclear whether this family had Sensenbrenner syndrome or a mild form of Jeune syndrome.
As in Sensenbrenner syndrome, genes associated with Jeune syndrome encode proteins involved in ciliary transport. Mutations have been identified in IFT80 (IFT 80; MIM 611177) that encodes an IFT-B complex protein, as well as in DYNC2H1 (dynein, cytoplasmic 2, heavy chain 1; MIM 603297), which is a retrograde motor transporting the IFT-A particle.31 32 Because of the clinical and functional overlap between Sensenbrenner and Jeune syndrome, we also performed mutation analysis in 17 patients with Jeune syndrome. Although we did not detect any C14ORF179 mutations in these patients, which indicates that this is not a frequent cause of Jeune syndrome, it remains a functional candidate gene.
In conclusion, we identified a homozygous mutation in the translation initiation codon in C14ORF179 that causes Sensenbrenner syndrome in a consanguineous family of Moroccan descent. The identified genetic defect interferes with translation, resulting in a shortened protein. Consistent with the disruption of a member of the IFT-A protein complex, fibroblasts from one of the affected siblings (II:2) show a typical IFT-A defect (ie, accumulation of IFT-B complex proteins in the ciliary tip). Our results demonstrate that Sensenbrenner syndrome results from defects in retrograde IFT.
Acknowledgments
We thank the Sensenbrenner and Jeune families for their participation. We also thank PL Beales, AFM Hoogeboom, BCJ Hamel, I Stolte-Dijkstra, D Doherty and other clinicians for supplying additional patients with Sensenbrenner syndrome and other ciliopathies. We thank R Boulouiz and A Bertoliavella for providing Moroccan control DNA samples, G Pazour for sharing the anti-IFT88 and anti-IFT57 antibodies and C Janke for the GT335 antibody. We thank T Merkx and S van der Velde-Visser for technical assistance and FMP Cremers, AI den Hollander, J Fransen, J te Riet and M Wijers-Rouw for helpful discussions.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
HHA, EMHFB, NVAMK and RR contributed equally to this work.
Funding This research was supported by grants from the Dutch Kidney Foundation (KJPB 0.009; to HHA), the European Community's Seventh Framework Programme FP7/2009 under grant agreement No 241955, SYSCILIA (to RR) and a grant from the Netherlands Organization for Scientific Research (NWO Vidi-91786396; to RR).
Competing interests None.
Patient consent Obtained. This includes written informed consent from the parents of the patients (II:1 and II:2) for publication of the images.
Ethics approval This study was conducted with the approval of the medical ethics committee of Radboud University Nijmegen Medical Centre.
Provenance and peer review Not commissioned; externally peer reviewed.