Article Text

Abstract
Background: The 9q subtelomeric deletion syndrome (9qSTDS) is clinically characterised by moderate to severe mental retardation, childhood hypotonia and facial dysmorphisms. In addition, congenital heart defects, urogenital defects, epilepsy and behavioural problems are frequently observed. The syndrome can be either caused by a submicroscopic 9q34.3 deletion or by intragenic EHMT1 mutations leading to haploinsufficiency of the EHMT1 gene. So far it has not been established if and to what extent other genes in the 9q34.3 region contribute to the phenotype observed in deletion cases. This study reports the largest cohort of 9qSTDS cases so far.
Methods and results: By a multiplex ligation dependent probe amplification (MLPA) approach, the authors identified and characterised 16 novel submicroscopic 9q deletions. Direct sequence analysis of the EHMT1 gene in 24 patients exhibiting the 9qSTD phenotype without such deletion identified six patients with an intragenic EHMT1 mutation. Five of these mutations predict a premature termination codon whereas one mutation gives rise to an amino acid substitution in a conserved domain of the protein.
Conclusions: The data do not provide any evidence for phenotype–genotype correlations between size of the deletions or type of mutations and severity of clinical features. Therefore, the authors confirm the EHMT1 gene to be the major determinant of the 9qSTDS phenotype. Interestingly, five of six patients who had reached adulthood had developed severe psychiatric pathology, which may indicate that EHMT1 haploinsufficiency is associated with neurodegeneration in addition to neurodevelopmental defect.
Statistics from Altmetric.com
In the last decade, several microdeletion and duplication syndromes emerged from routine screening of subtelomere regions by multiplex ligation dependent probe amplification (MLPA) and fluorescence in situ hybridisation (FISH).1 2 3 4 The chromosome 9q subtelomeric deletion represents one of the most common subtelomere deletions (6% of all).3 In addition, the chromosome 9q subtelomere deletion syndrome (9qSTDS) is also one of the first clinically recognisable telomeric syndromes.3 5 6 7 8 9 Affected individuals invariably have severe hypotonia with speech and gross motor delay. The facial features comprise micro- or brachycephaly, hypertelorism, synophrys or arched eyebrows, mid face hypoplasia, a short nose with upturned nares, a protruding tongue with everted lower lip and down turned corners of the mouth. Approximately half of affected individuals have congenital heart defects, primarily atrial or ventricular septal defects (ASD or VSD). A substantial part (10–20%) have epilepsy and/or behavioural and sleep disturbances. A variety of other major and minor eye, ear, genital and limb anomalies have been reported.7 8 9 We demonstrated that the syndrome is caused by haploinsufficiency of Eu-chromatin Histon Methyl Transferase 1 (EHMT1) (OMIM 607001), a gene involved in histone methylation.10 11 12 13 14 15 Since this finding, we have implemented the testing for the 9q subtelomeric deletion syndrome in a DNA diagnostic setting. This analysis includes a screen for small deletions by a multiplex MLPA approach and for intragenic EHMT1 mutations by direct sequencing of the gene in MLPA negative cases. To date, it is not yet known if and to what extent the size of the deletions or type of EHMT1 mutations are related to particular features or severity of the 9qSTDS. Therefore we have carefully studied the clinical and molecular characteristics of a cohort of newly identified cases with the 9qSTDS.
Patients and methods
Patients included in this study were referred by clinical geneticists from the Department of Human Genetics of the Radboud University Nijmegen Medical Centre and through collaborating centres. All patients referred had the core phenotype of the 9qSTDS comprising mental retardation, hypotonia and facial characteristics. The cohort included 16 patients with a known 9q deletion identified through routine subtelomere chromosome testing or whole genome array, who were referred for confirmation and/or further fine mapping of the deletion. In 14 cases the deletion was identified by routine subtelomeric MLPA (P070 MSRC Holland) or FISH (Vysis probe).1 2 In the other two, a deletion was identified through whole genome array (Affymetrix 500 k single nucleotide polymorphism (SNP) array and custom oligonucleotide array EmArrayCyto6000_version2). The second cohort comprised 24 patients with the typical manifestations of the syndrome but with normal routine MLPA/FISH results. The selection of patients for this cohort was carried out by two of us (TK and HGB) based on facial characteristics.
DNA was obtained from peripheral blood cells and extracted according to standardised procedures. MLPA and direct sequencing of the EHMT1 gene was carried out as described previously.12
We used the structure of the pre-SET and SET-domains (PDB-file 2rfi) to study the effect of the missense mutations on the three dimensional structure of the protein. The WHAT IF & Yasara Twinset (www.yasara.org) was used for analysis of the effects of the amino acid substitution towards the protein structure. A more extensive explanation of the methodologies and results is available online (http://www.cmbi.ru.nl/∼hvensela/EHMT1).
Results
Molecular analysis
The size of 16 novel deletions is schematically shown in fig 1. Deletion fine mapping was performed in 16 patients in whom presence of a 9q deletion had already been suggested, based on findings by routine telomere studies or genome-wide array approaches. In seven patients the deletion was initially identified by FISH (patients 8, 9, 10, 11, 12, 13 and 14), in another seven patients routine MLPA had been abnormal (patients 1, 2, 3, 4, 7, 15 and 16), and in two patients interstitial 9q deletions were found by microarray analysis—patient 5 by custom oligonucleotide array EmArrayCyto6000_version2, and patient 6 by Affymetrix 500 k SNP array. The breakpoint and sizes of the deletions show a heterogeneous distribution. The most common deletion was found in six patients and had a size between 700 kb and 1.3 Mb (figure 1). The largest one is 3.1 Mb in size extending from CACNA1B to OLFM1 (patient 2). The smallest one is 40 kb, comprising exon 11 to 25 of the EHMT1 gene exclusively (patient 6).

Schematic drawing of the 9q subtelomeric region and the different deletion sizes identified. Indicated on the right the patients numbered corresponding to table 1, that were shown to have the respective deletion. Relative positions of routine fluorescence in situ hybridisation (FISH) and P070 multiplex ligation dependent probe amplification (MLPA) probes are indicated.
Further deletion analysis followed by direct sequencing of EHMT1 was performed in 24 patients with normal routine MLPA/FISH results. No additional deletions were identified by MLPA whereas, in total, six novel intragenic mutations were found that are highly likely pathogenic (table 1). The mutations comprised two nonsense changes, c.778C>T and c.1717C>T (p.Arg260X, p.Gln573X), and one 4 bp deletion c.1440_1443del (p.Asp481fs), all three predicted to result in nonsense mediated mRNA decay (NMD). Two comprised de novo splice site mutations (c.2775-1G>A and c.2100-1G>C).
Clinical characteristics of patients with EHMT1 intragenic mutations and (interstitial) subtelomeric deletions
These mutations have not been previously reported as pathogenic mutations; they are not present as non-pathogenic variants (http://www.ncbi.nlm.nih.gov/SNP/). In three cases the expected de novo occurrence was confirmed by analysing DNA from both parents, whereas for three other cases with loss of function mutations, parents could not be analysed. Based on the prediction software for splice sites (http://www.fruitfly.org/seq_tools/splice.html, http://www.cbs.dtu.dk/services/NetGene2/, and http://www.genet.sickkids.on.ca/∼ali/splicesitefinder.html), the c.2775-1G>A and c.2100-1G>C changes cause aberrant splicing. Finally, one de novo missense change c.3125G>A (p.Cys1042Tyr) was found. This mutation affects a highly conserved cysteine residue located in the pre-SET protein domain (figure 2).

Domain structure of the EHMT1 protein and sequence identity of the pre-SET domain of EHMT1, the paralogous human gene EHMT2 (G9a), and the mouse and Drosophila orthologues. Note the highly conserved cysteine (arrow and underlined) that is mutated in patient 20. D. malenogaster, Drosophila malenogaster; H. sapiens, Homo sapiens; M. musculus; Mus musculus.
Protein modelling
Cysteine 1042 is located in a cysteine cluster surrounding three zinc ions. This cysteine/zinc cluster is important for the local structure of this particular region of the pre-SET domain. The cysteines make strong interactions with the zinc ions thereby placing the intervening loops in a correct position for dimer interactions (figure 3 and supplemental data http://www.cmbi.ru.nl/∼hvensela/EHMT1/). Mutation of cysteine 1042 into a tyrosine will abolish the strong cysteine/zinc interaction, causing a less stable domain. Moreover, the side chain of tyrosine will not fit at position 1042. Introduction of this residue will completely disturb the structure of the zinc/cysteine cluster and displace the residues in the loops that interact with the other monomer. The mutation will destroy the conformation of this domain that is needed for dimer-conta.
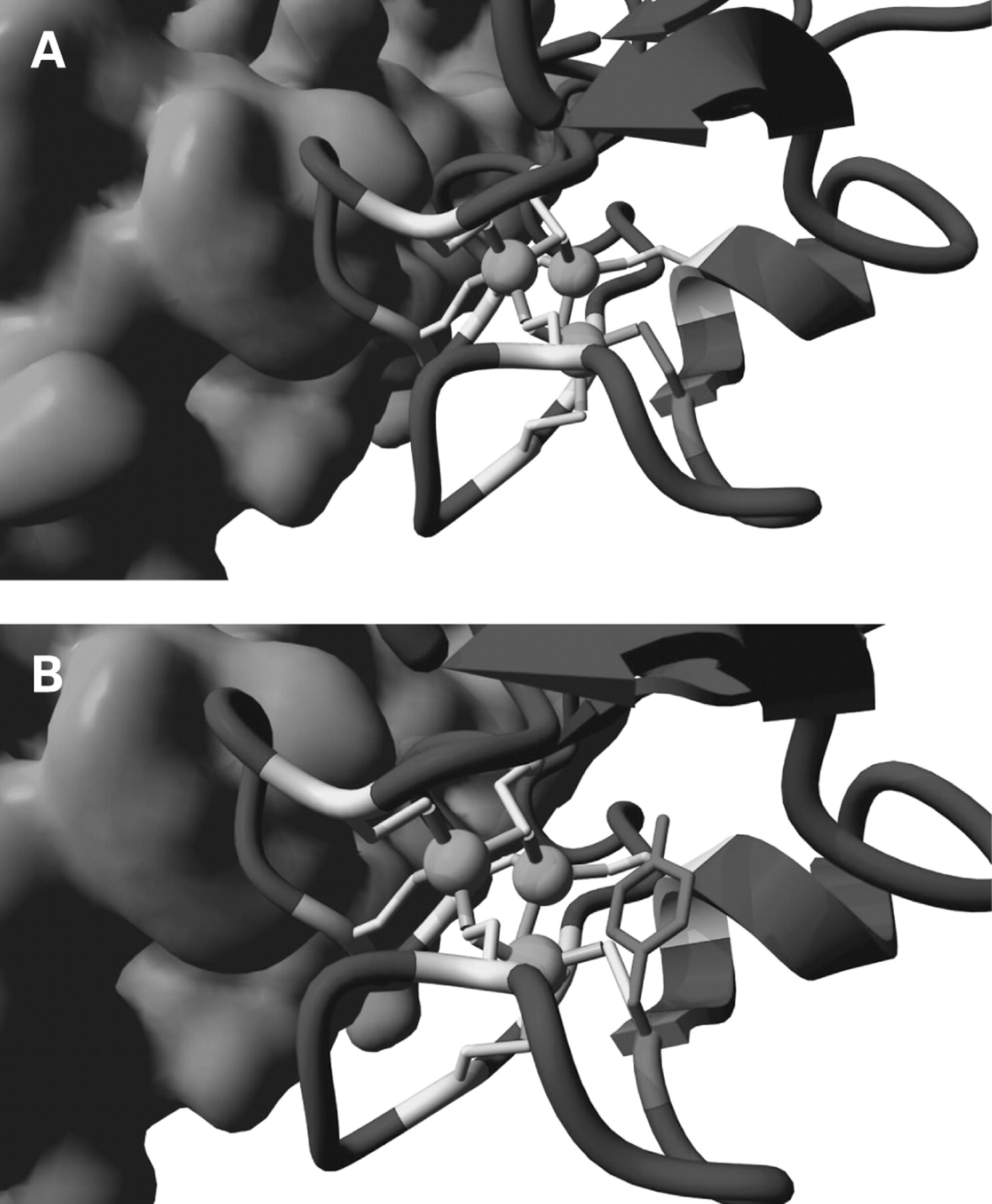
(A) Location of the Cys1042Tyr mutant. Cysteine 1042 is shown in red and is one of the cysteines in the cluster of cysteines (shown in yellow) that surround three zinc ions (green). Interactions of the cysteines with zinc are indicated as grey cylinders. The cluster is important for the correct position of the loops that interact with the other monomer (light blue surface). (B) Mutation of Cys1042Tyr. All cysteines in the cluster are coloured yellow, including cysteine 1042. This residue is mutated into a tyrosine (red); other colours are the same as the preceding figure. The side chain of tyrosine is shown to show the difference in size with the cysteine side chain.
Clinical data
Individual descriptions of patients are summarised in tables 1 and 2.
Developmental, behavioural, and psychiatric characteristics
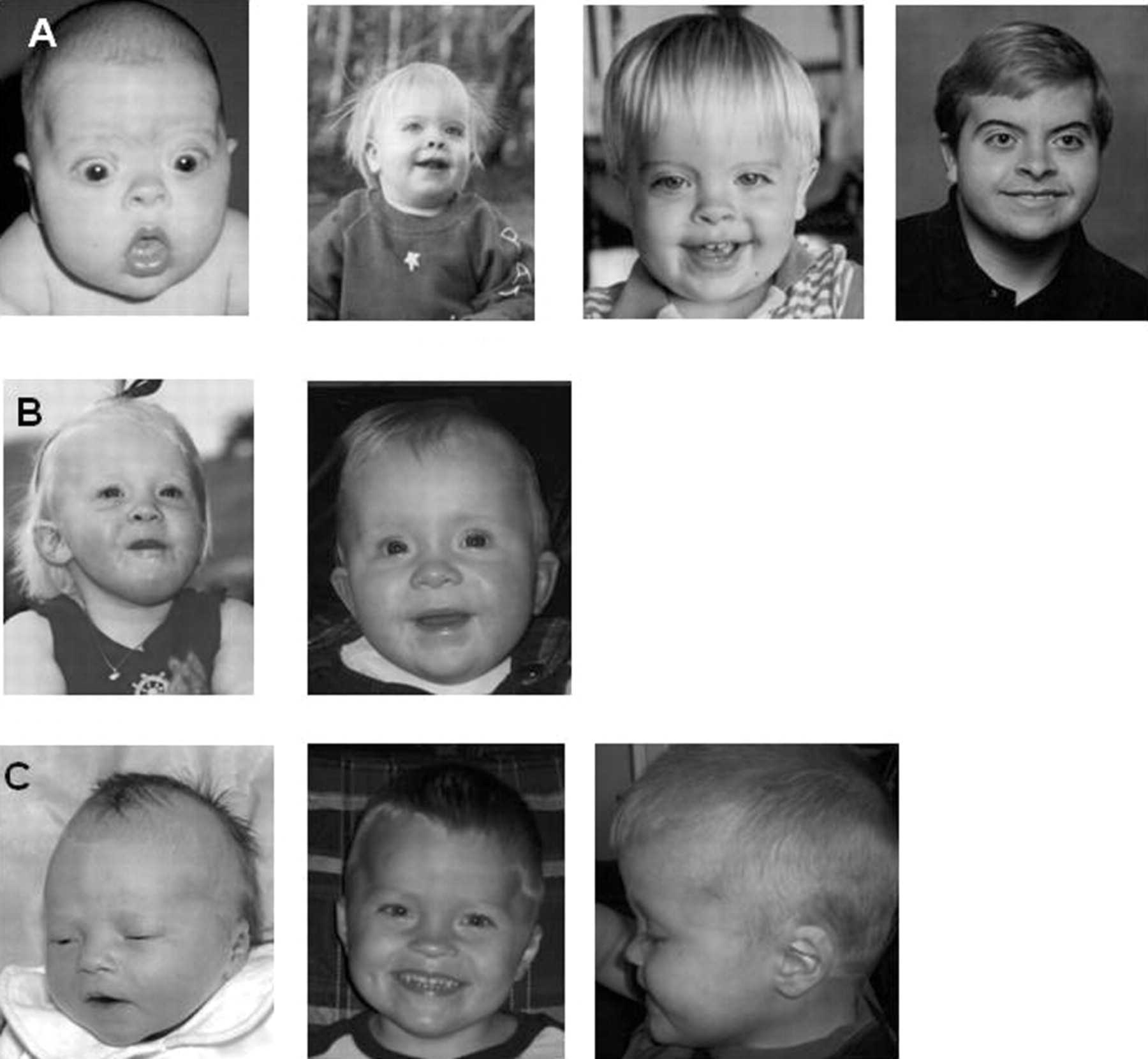
Clinical information of affected patients was systematically obtained by a standard clinical data sheet. No additional clinical data were obtained for patient 16. All patients showed the core phenotype of the 9qSTDS comprising mental retardation, childhood hypotonia, and recognisable facial features. Photographs showing the facial profile of several patients are shown in figures 4–7. Figure 4 shows five patients with an intragenic EHMT1 mutation. Figure 5 shows patients with obesity. When available and appropriate, series of photographs at different ages are depicted, showing the progression of the facial phenotype with age (figures 6 and 7).

Patients with EHMT1 mutations; patient 17 (A, B), patient 18 (C, ), patient 19 (E, F), patient 21 (G, H), patient 20 (I, J). Photographs of patients showing the characteristic facial profile comprising brachycephaly, hypertelorism, synophrys/arched eyebrows, mid face hypoplasia, protruding tongue, eversion of lower lip and prognathism of chin. Informed consent has been obtained for publication of all images present in this paper.

Patients 11 (A, B, C), 3 (D, E) and 15 (F, G) showing childhood obesity. Informed consent has been obtained for publication of all images present in this paper.

Patients at different ages showing evolution with age. Patient 12 (row A), patient 18 (row B), patient 13 (row C). Informed consent has been obtained for publication of all images present in this paper.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patients at different ages showing evolution with age. Patient 4 (row D), patient 8 (row E) and patient 7 (row F). Note asymmetric face in patient 8. Informed consent has been obtained for publication of all images present in this paper.
Birth weight in all patients was within or above normal range. In 4/15 deletion cases and 3/6 EHMT1 intragenic mutation cases, birth weight was at or above 90th centile. Childhood obesity was present in the same three mutated cases and one additional case (19) and in five deletion cases, noteworthy overlapping only with one who had high birth weight (patient 15). Microcephaly was noted in 6/15 patients with a deletion and 2/6 cases with a mutation and was already present at birth. No secondary microcephaly was reported. The degree of mental retardation was moderate to severe. Most did not develop speech, though language development was usually at a higher level making communication by other means, such as sign language or pictograms, possible. Variable cognitive function was noted. The best performance was in patient 11, with a deletion of minimal 970 kb, who can speak complete sentences (total intelligence quotient, TIQ = 52).
Motor function was delayed, but all individuals were able to walk, usually independently between 2–3 years of age. A notable exception is patient 6 who has an out of frame intragenic EHMT1 deletion (exons 11–25) and who is not able to walk at present age (7 years).
Variable congenital heart defects were observed in 7/15 deletion cases (ASD/VSD, tetralogy of Fallot, aortic coarctation, bicuspid aortic valve, and in patient 20 with the p.Cys1042Tyr EHMT1 mutation pulmonic stenosis). Genital defects were observed in 2/6 males with a deletion (micropenis, cryptorchidism) and in 1/2 males with a mutation (cryptorchidism). In addition vesico-ureteral reflux (VUR) was noted in patients 2 and 12 and chronic renal insufficiency was observed in patient 6 (intragenic EHMT1 deletion). Other anomalies were tracheo-/bronchomalacia with respiratory insufficiency and gastro-oesophageal reflux (GOR). Epilepsy was present in 6/15 deletion cases comprising tonic-clonic seizures, absences and complex partial epilepsy and generalised with focal onset in patient 19 with the p.Arg260X EHMT1 mutation. Results of brain imaging by CT or MRI performed in 12 individuals were generally normal but individual patients had corpus callosum hypoplasia (patient 22), enlarged cerebrospinal fluid (CSF) spaces/cortical atrophy (patients 5, 15, 19, 20), prominent Virchow–Robin spaces (patient 12), brain stem hypoplasia (patient 6), and abnormal myelinisation (patient 17).
Six patients reached adult age and were examined at 19, 20, 20, 33, 48, and 51 years of age, respectively. One of these died at age 19, probably because of cardiac arrhythmia, but autopsy was not performed. No behaviour characteristics were reported for this patient. In the other five adult patients an abrupt change in behaviour and daily functioning was observed starting at adolescence but recovered to some extent in adulthood. All five had been under psychiatric care and used psychotropic medication. Abnormalities included apathy, aggressive periods, psychosis or (autistic) catatonia, bipolar mood disorder, and regression in daily function and cognitive abilities.
Discussion
In this study the clinical and molecular data of 22 novel identified patients with the 9qSTDS are shown. Six of them had an intragenic EHMT1 mutation and the remaining 16 had submicroscopic 9q deletions of variable size, including seven interstitial deletions. The smallest deletion was 40 kb, taking out exons 11 to 25 of the EHMT1 gene exclusively. The seven interstitial deletions would have remained undetected by routine subtelomeric FISH analysis, since the probe that is commonly used for that purpose was not deleted. On the other hand, patient 15 showed abnormal FISH results, but unexpectedly, by validation with the standard MLPA probe (P070 in exon 9), normal results were found. It turned out that the proximal border of the deletion was disrupting the EHMT1 gene between exons 16 (not deleted) and 19 (deleted by MLPA). So neither routine subtelomeric testing with FISH nor with MLPA can exclude the presence of 9q deletions in all cases. Therefore, we recommend that testing with the more extensive 9q MLPA kit or high resolution genomic profiling should be used to discard definitely such deletions.
This study reports the smallest deletion reported so far, comprising only part of the EHMT1 gene (patient 6). Since it appeared that no other genes in the 9q region were involved, we considered this patient among the EHMT1 mutation cohort. This patient was known with tracheo-/bronchomalacia and chronic renal insufficiency with unknown cause in addition to the core phenotype. The largest deletion in this study extended into the TRAF2 gene where the most proximal MLPA probe was located. By additional SNP array, it was shown that the deletion was 3.1 Mb in size. This patient was born with several congenital abnormalities, encompassing micropenis, undescended testes, vesico-ureteral reflux, and aortic coarctation. It is possible that other genes in this larger deletion size are contributing to the severity of congenital defects seen in this patient; however, all of these features have also been observed in patients with smaller deletions or EHMT1 mutations.6 9 12 Moreover, a previously reported case with a 2.2 Mb interstitial 9q deletion, which completely overlaps the proximal part of the 3.1 Mb deletion described here, did not show any other major congenital abnormalities besides the mental retardation.11 So this does not support a major contribution of genes proximal of TRAF2 to the severity of the phenotype.
In six of 24 patients (25%) who were selected by us for exhibiting the 9qSTDS core phenotype, a pathogenic EHMT1 mutation was found. Five of six mutations disrupt the open reading frame of the EHMT1 gene and are predicted to create premature termination codons which may lead to nonsense mediated decay (NMD). Remarkably, one missense mutation was identified. Previously reported missense changes all turned out to be polymorphisms.12 However, the p.Cys1042Tyr missense change in the current study is most likely causal for the phenotype because of the de novo occurrence. Another argument is that the mutation replaces a highly conserved cysteine residue in the pre-SET domain of the Eu-chromatin Histon Methyl Transferase 1 protein. As predicted by the pre-SET domain model, this mutation will strongly influence the local conformation the pre-SET domain. The phenotype observed in this patient (patient 20) is fully compatible with the 9qSTDS. She had severe psychomotor retardation, hypotonia and characteristic facial profile with mid face hypoplasia, synophrys and eversion of the lower lip. It thus appears that the p.Cys1042Tyr change has a dramatic effect on the protein function, and may reflect an EHMT1 null-allele just like mutations in the other patients. A dominant negative effect is not expected since complete absence of the protein is not believed to be compatible with life.15 Further functional studies could give additional proof for pathogenicity of this mutation.
Another novel category among the mutations are the two splice site changes. Both are predicted to result in aberrant splicing and, consequently, disruption of the open reading frame. There were no cells available to test this further.
No significant differences were noted between previously reported patients with 9q subtelomeric deletions and patients with EHMT1 mutations (tables 1 and 3), except for high birth weight and childhood obesity.
However, the frequency of features such as birth weight and behaviour/psychiatric aspects are not consistently reported in previous studies. In addition to the previously recognised childhood obesity that is present in a higher frequency in the 9qSTDS,6 it seems that birth weight tends to be higher as well. When considering also previously reported data, high birth weight has been reported in approximately 10% of patients with small 9q deletions and in 50% of patients with EHMT1 mutations (table 3). The prevalence of heart defects is 30% in both cohorts. One patient with the p.Cys1042Tyr had a pulmonic stenosis, one previously reported patient with the p.Arg1137X had a VSD, and the originally reported patient with translocation breakpoint through EHMT1 had an ASD.10 12 Another patient with a small deletion of 200 kb, comprising only EHMT1 gene and possibly ARRDC1, had a tetralogy of Fallot.
Electronic database information
Online Mendelian Inheritance in Man (chromosome 9q subtelomere deletion syndrome 610253; EHMT1 607001): http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = OMIM&cmd = Limits
Unique (Rare Chromosome Disorder Support Group in the United Kingdom): http://www.rarechromo.org/html/home.asp
University of Nijmegen (clinical EHMT1 sequencing and MLPA screening): http://www.humangenetics.nl/en/diagnostics_en.php
Emory University (clinical EHMT1 sequencing): http://www.genetics.emory.edu/testing/index.php
University of California Santa Cruz (UCSC) Genome Browser (Assembly March 2006): http://genome.ucsc.edu
3-D EHMT1 (pre-)SET domain structure and Cys1042Tyr mutation: http://www.cmbi.ru.nl/∼hvensela/EHMT1/
Seizures were present in 20% (mutation cohort) and in 30% (deletion cohort), presenting in infancy or adolescence with tonic-clonic insults, absences or grand mal. Epilepsy can present severely in the first year of life or later in childhood. Other features mentioned were recurrent ear, nose and throat infections (eight patients), persistence of deciduous teeth (five patients), pigmentation defect (one patient), hearing loss (mixed or perceptive in six patients), and pes equinovarus (one patient). A variable spectrum of behavioural abnormalities was observed in both patient categories. Frequently reported behavioural problems in younger children are temper tantrums, severe sleep disturbances, and autistic features. However, sociable skills, friendliness and happiness were also mentioned. A sudden and dramatic change in behaviour and performance was observed during adolescence in five of the six adult patients leading to psychiatric problems. These patients were diagnosed with psychosis, bipolar mood disorder and/or autistic catatonia. Noteworthy, in two of them a transient or even permanent loss of functioning with loss of skills as toilet training and the ability to independent eating occurred. Disturbance of epigenetic gene regulation can result in regression of daily performance and cognitive abilities as demonstrated in Rett syndrome (MIM 312750). Regression is one of the hallmarks of this condition that is caused by disturbance of chromatin modulation, through mutations in the Methyl-CpG- Binding Protein 1 (MeCP2) gene.16 It has been shown that the MeCP2 protein thereby interacts with several interesting proteins, as the ATRX protein, a SWI2/SNF2 DNA helicase/ATPase,17 and histone methyl transferase Suv39H1.16
To our knowledge, there is as yet no direct evidence as that any of these proteins directly interact with EHMT1. However, it has been demonstrated that Suv39H1 is found in complexes with the paralog of EHMT1, the G9a protein,18 which in turn forms functional complexes with the EHMT1 protein.15 This raises the question whether haploinsufficiency of EHMT1 similarly leads to regression in function and behaviour pattern, albeit at a later age. Further detailed neuropsychiatric studies are needed to get more insight into these features. Such information clearly will be of great importance to parents and caregivers of 9qSTDS patients.
REFERENCES
Footnotes
For numbered affiliations see end of article
Competing interests None declared.
Patient consent Obtained.
Provenance and Peer review Not commissioned; externally peer reviewed.