Article Text

Abstract
Background: Pulmonary arterial hypertension (PAH) is a progressive disorder characterised by raised pulmonary artery pressures with pathological changes in small pulmonary arteries. Previous studies have shown that approximately 70% of familial PAH and also 11–40% of idiopathic PAH (IPAH) cases have mutations in the bone morphogenetic protein receptor type II (BMPR2) gene. In addition, mutations in the activin receptor-like kinase 1 (ALK1) gene have been reported in PAH patients. Since both the BMPR2 and ALK1 belonging to the transforming growth factor (TGF)-β superfamily are known to predispose to PAH, mutations in other genes of the TGF-β/BMP signalling pathways may also predispose to PAH.
Methods: We screened for mutations in ENDOGLIN(ENG), SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD6 and SMAD8 genes, which are involved in the TGF-β/BMP signallings, in 23 patients with IPAH who had no mutations in BMPR2 or ALK1.
Results: A nonsense mutation in SMAD8 designated c.606 C>A, p.C202X was identified in one patient. The father of this patient was also identified as having the same mutation. Functional analysis showed the truncated form of the SMAD8 C202X protein was not phosphorylated by constitutively active ALK3 and ALK1. The SMAD8 mutant was also unable to interact with SMAD4. The response to BMP was analysed using promoter-reporter activities with SMAD4 and/or ca-ALK3. The transcriptional activation of the SMAD8 mutant was inefficient compared with the SMAD8 wild type.
Conclusion: We describe the first mutation in SMAD8 in a patient with IPAH. Our findings suggest the involvement of SMAD8 in the pathogenesis of PAH.
Statistics from Altmetric.com
Pulmonary arterial hypertension (PAH; MIM 178600) is a rare condition, with an annual incidence of 1–2 cases per 1 million in the general population.1 2 It is characterised by abnormal proliferation of endothelial and smooth muscle cells in the pulmonary arterioles, which leads to sustained elevation of mean pulmonary artery pressure ⩾25 mm Hg at rest and/or ⩾30 mm Hg during exercise.1 3 Although the disease may present at any age, PAH is usually diagnosed in the fourth decade of life, with a female-to-male ratio of 2 to 1.1 3 Without modern treatments the disorder progresses rapidly, leading to right heart failure within 3 years of diagnosis. It has been reported that among all PAH patients, familial PAH accounts for at least 6% of the cases, with 10–20% of penetrance.1 3
A recent classification study proposed five subgroups of PAH: idiopathic PAH (IPAH); familial PAH (FPAH); PAH associated with other disease (APAH) such as collagen vascular disease, congenital systemic to pulmonary shunts, portal hypertension, and HIV infection; PAH associated with significant venous or capillary involvement; and persistent pulmonary hypertension of the newborn (PPHN).4 5
A study in France reported that FPAH and IPAH accounted for prevalence rates of 4.2% and 38.9% in 553 PAH cases, respectively.6 FPAH and IPAH share the same clinical feature, histopathology and clinical course, and FPAH has a low penetrance. Therefore, the true incidence and prevalence of FPAH have not been fully elucidated.7
Heterogeneous germline mutations in the bone morphogenetic protein (BMP) receptor type II gene (BMPR2), a receptor for the transforming growth factor (TGF)-β superfamily on chromosome 2q33, have been identified in FPAH.8 9 BMPR2 mutations have been identified in up to 70% of FPAH and 11–40% of patients with IPAH.10 11
Mutations in the activin receptor-like kinase 1 (ALK1) gene, a member of the TGF-β superfamily, on chromosome 12q13 have also been reported in IPAH/FPAH patients12 13 and in patients with hereditary haemorrhagic telangiectasia (HHT) associated PAH.14 15 Except for the association with BMPR2 and ALK1 mutations, there is a paucity of published studies reporting broad and systematic mutation screening of PAH patients.12 16 Because both BMPR2 and ALK1 genes, which belong to the TGF-β superfamily, are known to predispose to PAH, the question is raised as to whether mutations in other genes of this signalling pathway may also predispose to PAH. In this study, 23 patients clinically diagnosed with IPAH, who had no mutations in BMPR2 or ALK1, were screened for mutations in eight genes involved in the TGF-β/BMP signalling pathway—ENDOGLIN (ENG), SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD6 and SMAD8 (which also known as SMAD9)—to determine whether any of them are involved in the pathogenesis of PAH.
METHODS
Subjects
All assessments were done with the approval of the ethics committees of Tokyo Women’s Medical University and Toho University, Tokyo, Japan. Patients were recruited from Tokyo Women’s Medical University and Toho University. Written informed consent was obtained from all study subjects. If patients were under 16 years of age, the informed consent was given by their guardians. We assessed each patient by clinical history, physical examination and a review of their medical records.
The diagnosis of IPAH/FPAH was made through clinical evaluation, chest radiography, electrocardiography, echocardiography and cardiac catheterisation based on current international consensus criteria (Venice 2003).5 6 Patients with APAH, PAH associated with significant venous or capillary involvement, PPHN and other pulmonary hypertensions were excluded from this study by trained cardiologists.
This study contains individuals clinically diagnosed with IPAH who were derived from two cohorts. The first cohort of 12 patients with IPAH was derived from a previous study13 and these 12 patients were confirmed to be without the BMPR2 or ALK1 mutation by direct sequencing and multiplex ligation dependent probe amplification (MLPA) analysis.11 The second cohort contained 21 patients with either IPAH or FPAH, and mutation analysis revealed 10 patients, nine of whom had mutations inclusive of three patients with exonic deletions in BMPR2, and one had a mutation in ALK1 (unpublished data). These 10 patients were excluded from this study. The remaining 11 patients with IPAH from the second cohort and the 12 patients with IPAH from the first cohort, 23 patients in total, who were confirmed to have no mutation in BMPR2 or ALK1, were included in this study.
Summary of baseline characteristics and haemodynamic parameters of the 23 patients is presented in table 1. Available data on characteristics and haemodynamic parameters of 23 patients with IPAH are provided in supplemental table 1.
Sequencing and mutation analysis
Genomic DNA was extracted from peripheral blood leucocytes of the patients or from their lymphoblastoid cell lines transformed by the Epstein–Barr virus as described previously.17 In the 23 patients with no mutations in BMPR2 or ALK1, all coding exons and adjacent intronic regions for ENG, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD6 and SMAD8 were amplified using polymerase chain reaction (PCR primer details are available in supplemental table 2). PCR amplified products were purified and directly sequenced as previously described.18
In all cases, any sequence variations found were reamplified and resequenced to confirm the observed changes. The observed changes were also confirmed by amplification refractory mutation system (ARMS) assay.19
The sequences generated were compared with wild type ENG (GenBank accession number NM_000118), SMAD1 (GenBank NM_005900), SMAD2 (GenBank NM_005901), SMAD3 (GenBank NM_005902), SMAD4 (GenBank NM_005359), SMAD5 (GenBank NM_005903), SMAD6 (GenBank NM_005585), and SMAD8 (GenBank NM_005905).
When a mutation was detected, we confirmed that it was not present in 150 healthy controls by direct sequencing.
Plasmids and antibodies
Human pcDNA3.0-6xMyc-SMAD8, human pcDNA3.0-Flag-SMAD4, human pcDNA3.0-ALK1-haemagglutinin (HA), human pcDNA3.0-ALK3-HA, BMP-responsive promoter reporter construct 3GC2-Lux, and TGF-β responsive promoter reporter construct 3TP-Lux were kindly provided by Dr K Miyazono (Tokyo, Japan). 3GC2-Lux contains three repeats of a GC-rich sequence derived from the proximal BMP response element in the Smad6 promoter.20 3TP-Lux is a TGF-β responsive luciferase reporter gene that contains three consecutive tetradecanoylphorbol acetate (TPA) response elements and a portion of the plasminogen activator inhibitor 1 (PAI-1) promoter region.21
Human constitutively active (ca) ALK1 and ALK3 were generated by mutation of Glu-201 into aspartic acid and a mutation of Glu-233 into aspartic acid, respectively.
Site directed mutagenesis was carried out by a PCR based approach. The constructed plasmids were verified by sequencing. The antibodies used were as follows: anti-Flag antibody (F3165, Sigma, St Louis, Missouri, USA), anti-HA antibody (11867423001, Roche, Mannheim, Baden-Württemberg, Germany), anti-Myc antibody (#2276, Cell Signaling Technology, Danvers, Massachusetts, USA), anti-Myc antibody (#06-549, Upstate, Lake Placid, New York, USA) and anti-phospho-Smad1/Smad5/Smad8 antibody (#9511, Cell Signaling Technology).
Transfection, cell lysis, immunoblotting and immunoprecipitation
HEK293 and COS1 cells were grown in DMEM/F-12 (Sigma) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, New York, USA), 100 units/ml penicillin/streptomycin (Gibco) and 250 ng/ml amphotericin B (Sigma). Transfection was performed with Lipofectamine 2000 reagent (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were lysed in lysis buffer (1M Tris-HCl (pH 8.0) 50 mM, 0.5 M EDTA 1 mM (pH8.0), 5 M NaCl 120 mM, NP-40 0.25%). For co-immunoprecipitation assays, the lysates were incubated with monoclonal anti-Flag antibody (Sigma) and protein G-Sepharose beads (GE Healthcare, Little Chalfont, Buckinghamshire, UK), and immunoblotted with polyclonal anti-Myc antibody (Upstate).
Luciferase assay
COS1 cells were co-transfected in Opti-MEM (Invitrogen) using Lipofectamine 2000 reagent (Invitrogen) with 3GC2-Lux or p3TP-Lux and wild type or mutant pcDNA3.0-SMAD8 and/or pcDNA3.0-SMAD4 and/or pcDNA3.0-ca-ALK3 (total 0.9 μg). Twenty-four hours after transfection, the cells were harvested. Firefly and renilla luciferase activities were measured with the Dual luciferase reporter assay (Promega, Madison, Wisconsin, USA) following manufacturer’s instructions. Results were expressed as the ratio of firefly luciferase activity to renilla luciferase activities. All assays were performed in triplicate.
Statistics
All results are expressed as mean (SD). For statistical comparison of two samples, a two-tailed Student’s t test was used where applicable. Values of p<0.05 were considered significant.
RESULTS
Sequence analysis
To identify mutations in genes involved in the TGF-β/BMP signalling pathway, we screened mutations in ENG, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD6 and SMAD8 genes in 23 patients with IPAH who had no mutations in BMPR2 or ALK1.
In this study, no mutations were identified in ENG, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5 or SMAD6, whereas common polymorphisms were found in ENG, SMAD3 and SMAD6 (supplemental table 3).
However, a nonsense mutation in SMAD8, c.606 C>A, p.C202X was identified in one patient (proband 14) (fig 1A, B). The SMAD8 mutant introduces a premature stop codon into exon 2 and results in a truncated protein that lacks 228 carboxy-terminal amino acids, including the MH2 domain and the SXS phosphorylation site (fig 1C).

SMAD8 mutation in pulmonary arterial hypertension. (A) DNA sequences showing c.606 C>A in SMAD8. (B) The panel shows confirmation of the mutation by ARMS assay using a reverse primer specific for the mutant allele yields a 330 bp product but no product in a control individual. The upper band is of the internal control. (C) Schematic representation of SMAD8 wild type and SMAD8 C202X mutant. (D) Pedigree of the patient’s family. PAH, pulmonary arterial hypertension.
Clinical characteristics
The patient harbouring a mutation in SMAD8 was diagnosed with PAH based on an accentuated second heart sound in the pulmonary region during hospitalisation with pneumonia at 8 years of age. His haemodynamic data at 8 years of age showed a mean pulmonary arterial pressure (mPAP) of 53 mm Hg, right atrial pressure (RAP) of 5 mm Hg, cardiac index (CI) of 4.31 litres·min−1·m−2, total pulmonary resistance (TPR) of 12.3 Wood·Unit−1·m−2, pulmonary vascular resistance (PVR) of 12.4 Wood·Unit−1·m−2 and pulmonary artery wedge pressure (PAWP) of 6 mm Hg. His condition progressed to World Health Organization functional class III with episodes of shortness of breath during physical activity at 9 years of age. He has been receiving epoprostenol, continuous intravenous prostacyclin, since the age of 9.
His current condition is WHO functional class I–II at 16 years of age. His recent haemodynamic data showed mPAP of 49 mm Hg, RAP of 7 mm Hg, CI of 4.98 litres·min−1·m−2, TPR of 9.9 Wood·Unit−1·m−2, PVR of 8.5 Wood·Unit−1·m−2 and PAWP of 7 mm Hg.
The patient’s mother and his elder brother did not have the same mutation. However, his 58-year-old father was identified as having the same mutation although no clinical symptoms of PAH were observed (supplemental fig 1). According to the patient’s father, he was the sixth of seven children. His third sister and fifth brother died of pulmonary diseases at 13 years of age and at ⩽2 years of age, respectively (fig 1D). The other four siblings of the father have not been screened for SMAD8 mutations, because their blood samples were not obtainable at this point in time.
Immunoblotting assay and co-immunoprecipitation
SMAD8 mutant was neither phosphorylated nor stimulated to interact with SMAD4 by constitutively active ALK3 and ALK1
It has previously been reported that Smad8 is phosphorylated by constitutively active TGF-β/BMP type I receptors, except ca-ALK5, and is stimulated to interact with Smad4.22 23 Moreover, a C-terminal truncated Smad8 lacking the SXS phosphorylation site was shown to be unphosphorylated in the presence of ca-ALK3 and failed to interact with Smad4.22
We first examined the effects of ca-ALK3 and ca-ALK1 on the phosphorylation of the SMAD8 C202X mutant. The SMAD8 mutant was not phosphorylated in the presence of ca-ALK3 and ca-ALK1 (fig 2A). We further examined the ability of the SMAD8 mutant to interact with SMAD4 by co-immunoprecipitation. Interaction between SMAD8 wild type and SMAD4 was observed in the presence of ca-ALK3 or ca-ALK1. However, the SMAD8 mutant failed to interact with SMAD4 (fig 2B).

SMAD8 C202X mutant is not phosphorylated by TGF-β/BMP type I receptors, and does not interact with SMAD4. (A) Phosphorylation of Myc-SMAD8 wild type and SMAD8 C202X mutant in COS1 cells were examined by immunoblotting (Blot). Myc-SMAD8 wild type or SMAD8 C202X mutant was transiently co-expressed with constitutively active HA-TGF-β/BMP type I receptor ALK3 and ALK1. The type I receptor dependent phosphorylation of SMADs was shown by anti-phospho-Smad1/5/8 antibody, which also reacts with phosphorylation of SMAD8. (B) The interactions of Myc-SMAD8 wild type and SMAD8 C202X mutant with Flag-SMAD4 in HEK293 cells were examined by immunoprecipitation (IP) followed by immunoblotting (Blot). HEK293 cells were transfected with SMAD8 wild type, SMAD8 C202X mutant, SMAD4 and constitutively active ALK3 and ALK1. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody and analysed by immunoblotting with anti-Myc antibody.
Luciferase assay
SMAD8 mutant was not capable of activating the BMP/TGF-β responsive promoter-reporter
It has been reported that Smad8 alone or co-expressed with Smad4 increases BMP responsive promoter-reporters in the absence of either BMP ligands or constitutively active TGF-β/BMP type I receptors.23 Therefore, to determine if the SMAD8 C202X mutant is able to increase BMP and TGF-β responsive promoter-reporter activity, we investigated the transcriptional activity mediated by SMAD8 wild type and the mutant with or without SMAD4 and/or ca-ALK3 by using the BMP responsive promoter-reporter, 3GC2-Lux and the TGF-β responsive promoter-reporter, 3TP-Lux in COS-1 cells.
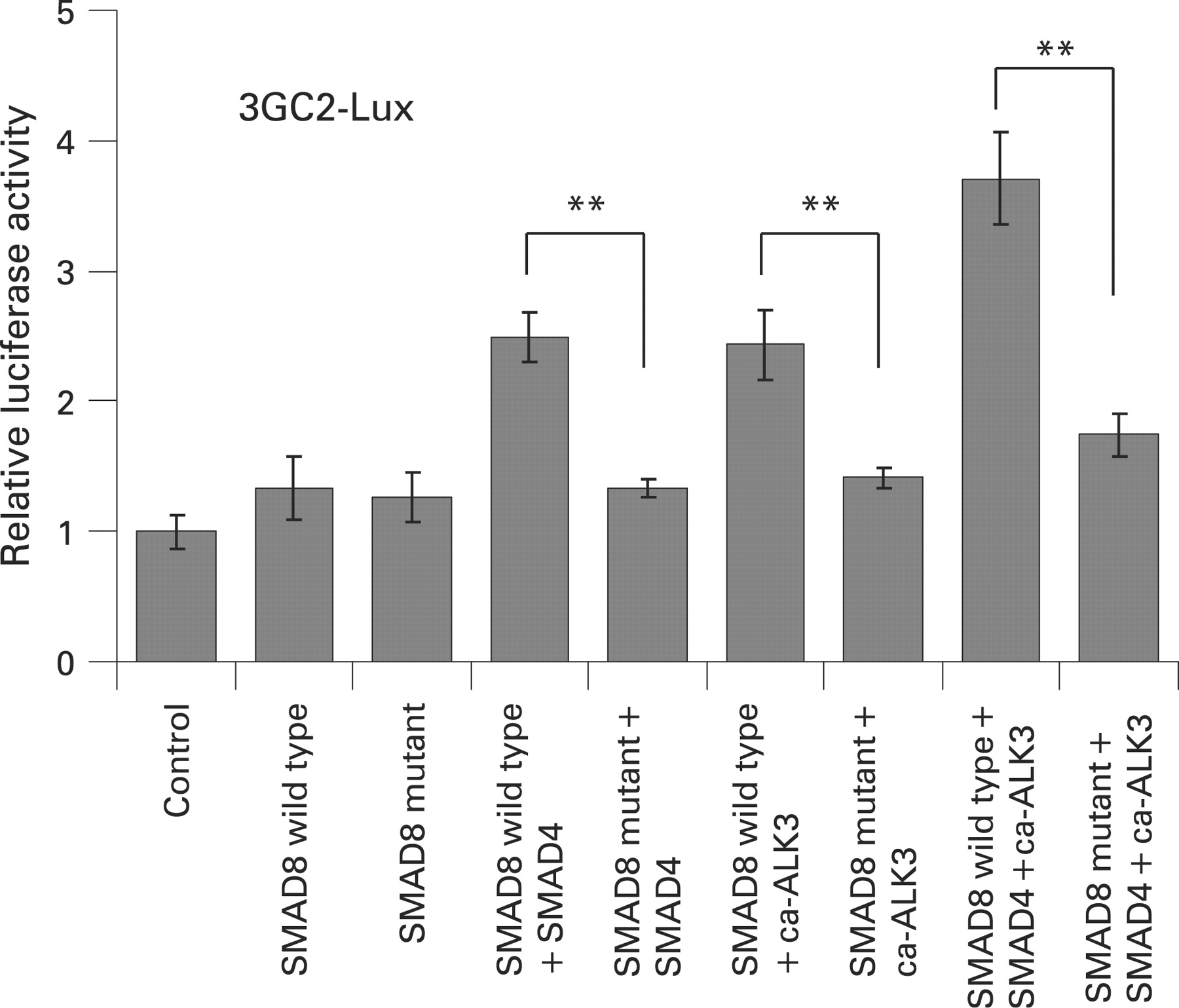
SMAD8 wild type induced BMP responsive promoter activity. The co-expression of SMAD8 wild type with either SMAD4 or ca-ALK3, induced approximately two times higher activity than SMAD8 wild type alone. The SMAD8 wild type co-expressed both SMAD4 and ca-ALK3 induced approximately three times higher activity than the wild type alone. However, the SMAD8 mutant, even when co-expressed with SMAD4 or/and ca-ALK3, were inefficient in activating the BMP responsive promoter-reporter compared with SMAD8 wild type (fig 3).

{kind=link}
{kind=link}
{kind=link}
The effect of SMAD8 wild type and SMAD8 C202X mutant on the transcriptional activation of 3GC2-Lux were determined in the presence and absence of SMAD4 and/or ca-ALK3 in COS1 cells. Values represent mean (SD). Statistical differences between groups were assessed by Student’s t test. **p<0.01.
Similar results were obtained using the TGF-β responsive promoter-reporter, 3TP-Lux (supplemental fig 2). These results demonstrate that the SMAD8 C202X mutant is not capable of activating BMP/TGF-β responsive promoter-reporters.
DISCUSSION
Smad8 is one of the receptor regulated Smads (R-Smads) for the TGF-β superfamily of receptors. Smad8 as well as Smad1 and Smad5 are direct substrates of TGF-β/BMP type I receptors and are phosphorylated at the SXS motif in the C-terminal region. Phosphorylated Smad8 then associates with Co-Smad or Smad4 and translocates into the nucleus, where they regulate the transcription of target genes.24
Previous reports have suggested that the loss of signals mediated by SMAD1/5/8 plays an important role in pulmonary vascular remodelling and the pathogenesis of PAH. Reduced phosphorylation of SMAD1 within the media and intima cells of small pulmonary arteries in patients with IPAH and FPAH was observed.25 The expression of BMPR2 in pulmonary vascular lesions of IPAH/FPAH patients, especially in those having the BMPR2 mutation, was reduced.26 Functional studies on BMPR2 mutations have shown that BMPR2 mutations disrupted or down regulated SMAD1/5/8 signalling.27 28 The expression of ALK3 was markedly reduced in the lung of IPAH patients who had no mutations in BMPR2 or ALK1.29 Furthermore, expression of phosphorylated Smad1/5/8 in lung was markedly reduced in conditional knockout mice lacking Alk3 in alveolar epithelium.30 Functional analysis of ALK1 mutations in HHT have shown that ALK1 mutations disrupt or down regulate SMAD1/5/8 signalling.31 These observations indicate that the down regulation of TGF-β/BMP signals via SMAD1/5/8 plays an important role in the pathogenesis of PAH.
BMPR2 has been identified as the major causative gene of IPAH/FPAH.8–10 Also, ALK1 mutations have been reported in patients with IPAH/FPAH.12 13 A ENG mutation was reported in an infant patient with IPAH and was later diagnosed with HHT at 8 years of age.12 However, approximately 30% of FPAH and 60–89% of apparently IPAH patients do not harbour mutations in BMPR2.10 11 About 70% of paediatric patients with IPAH do not harbour mutations in either BMPR2 or ALK1.13 Therefore, in this study, we investigated whether mutations in other genes involved in TGF-β/BMP signalling pathways occur in 23 patients with IPAH.
We identified a nonsense mutation in SMAD8, c.606 C>A, p.C202X in one patient (fig 1A). To our knowledge, a germline mutation of SMAD8 has not yet been reported in either PAH or other diseases, whereas loss of SMAD8 expression in breast, colon and prostate cancer has been reported.32 33
As predicted by the truncated protein structure of the SMAD8 C202X mutant (fig 1C), our immunoblotting and co-immunoprecipitation assay showed that it was not phophorylated by TGF-β/BMP type I receptors, and that it did not interact with SMAD4 (fig 2A, B). These results indicate the SMAD8 mutant disturbs the downstream signalling of TGF-β/BMP.
It has previously been reported that Smad8 was able to translocate into the nucleus in the absence of ca-ALKs, although its translocation efficiency was much less than that observed in the presence of the ca-ALKs.23 Therefore, we investigated whether the SMAD8 mutant could perform some of the transcription functions of the wild-type protein by using a BMP responsive promoter-reporter, the 3GC2-Lux construct, in COS-1 cells. The luciferase assay showed that SMAD8 wild type produced a highly significant increase in BMP responsive promoter-reporter activity, particularly when it was transfected with both SMAD4 and ca-ALK3. In contrast, the SMAD8 mutant was inefficient in activating the reporter expression with either SMAD4 or ca-ALK3 (fig 3). This can be explained by the truncated protein structure of the SMAD8 mutant which lacks both the MH2 domain and the SXS phosphorylation site in the C-terminal region. The MH2 domain is not only required for SMAD4 interaction but also for binding to other nuclear factors such as DNA binding cofactors, co-activators and co-repressors for the assembly of transcriptional complexes.24
The clinical phenotype of our patient with SMAD8 mutation was not different from other IPAH patients. He has no other symptoms other than those of PAH at 16 years of age. His father has the same SMAD8 mutation, but clinical symptoms or objective measurement diagnostics of PAH have not been observed to date. This situation is not surprising as FPAH with germline BMPR2 mutations has a low penetrance. Only 10–20% of BMPR2 mutation carriers will manifest clinical PAH.9 34 Low penetrance was also observed in our previous study; two fathers of paediatric patients with FPAH had the same ALK1 mutation as their children, but had no symptoms of PAH or HHT.13
In this study, a SMAD8 mutation was found in only one patient out of the 23 IPAH patients (1/23, 4.3%). Clearly, additional IPAH/FPAH patients will have to be screened to determine the prevalence, penetrance and precise role of SMAD8 in IPAH/FPAH.
Previous studies have suggested that the down regulation of TGF-β/BMP signals via SMAD1/5/8 plays an important role in the pathogenesis of PAH. Our finding of SMAD8 mutation in a PAH patient provides one more piece of evidence to support it.
The 22 of the 23 patients with IPAH (22/23, 95.7%) were not identified mutations in ENG, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD6 and SMAD8 in this study. It suggests that there are still unidentified genes in the TGF-β/BMP signal pathways or other pathways predisposing to PAH. In addition, the low penetrance of FPAH despite the mutation in broadly expressed genes indicates that additional genetic and/or environmental factors play a critical role in the development of PAH in carriers of the SMAD8, BMPR2 and ALK1 mutations. It has been reported that prolonged hypoxia induces pulmonary vascular remodelling and PAH in mice.35 BMPR2 heterozygous mutant mice responded to inflammatory stress with a marked increase in right ventricular systolic pressure.36 Thus, it will be interesting to determine whether Smad8 homozygous or heterozygous mutant mice develop PAH in response to stimuli such as chronic hypoxia or inflammatory stress. Furthermore, to determine the functions of SMAD8 in the context of PAH, further investigations using human pulmonary artery smooth muscle cells and human pulmonary artery endothelial cells will be necessary.
Acknowledgments
We are grateful to the patients and their family members. We thank Dr Bernardo Nadal-Ginard for his valuable comments. We thank Dr Kohei Miyazono for providing the plasmids. We also thank Dr Shin-ichiro Imamura, Dr Shoichi Arai, Dr Yoshiyuki Furutani, Dr Maya Fujiwara, Dr Emiko Hayama and Ms Michiko Furutani for their excellent technical assistance.
REFERENCE
Supplementary materials
Web only appendices 46:5;331-7
Files in this Data Supplement:
Footnotes
▸ Additional tables and figures are published online only at http://jmg.bmj.com/content/vol46/issue5
Funding: This work was supported by the Program for Promoting the Establishment of Strategic Research Centers, Special Coordination Funds for Promoting Science and Technology, Ministry of Education, Culture, Sports, Science and Technology (Japan).
Competing interests: None.
Patient consent: Obtained.