Article Text

Abstract
This report presents the detection of a heterozygous deletion at chromosome 12q14 in three unrelated patients with a similar phenotype consisting of mild mental retardation, failure to thrive in infancy, proportionate short stature and osteopoikilosis as the most characteristic features. In each case, this interstitial deletion was found using molecular karyotyping. The deletion occurred as a de novo event and varied between 3.44 and 6 megabases (Mb) in size with a 3.44 Mb common deleted region. The deleted interval was not flanked by low-copy repeats or segmental duplications. It contains 13 RefSeq genes, including LEMD3, which was previously shown to be the causal gene for osteopoikilosis. The observation of osteopoikilosis lesions should facilitate recognition of this new microdeletion syndrome among children with failure to thrive, short stature and learning disabilities.
- FISH, fluorescence in situ hybridisation
- Mb, megabase
- osteopoikilosis
- short stature
- mental retardation
- HMGA2
- GRIP1
Statistics from Altmetric.com
Classical cytogenetic analysis has played an essential role in the discovery of recurrent segmental deletions in patients with clinically recognisable mental retardation such as Prader–Willi, Miller–Dieker, Langer–Giedion and velocardiofacial syndromes.1 The subsequent delineation of commonly deleted segments and mapping of small atypical deletions have allowed the identification of genes responsible for the major clinical features of these contiguous gene deletion syndromes.2 Recently, molecular karyotyping was proven to be a more powerful tool in detecting submicroscopic deletions or duplications in patients with so-called idiopathic mental retardation with or without congenital malformations. Molecular karyotyping studies have shown that in about 10% of these cases segmental imbalances can be found.3–9 Using this new genome-wide screening technology, new disease genes can be identified, as illustrated for the CHD7 gene in CHARGE syndrome.10 Very recently, some new microdeletion syndromes were identified using molecular karyotyping.11–13
Here, we report on three unrelated patients with de novo 12q14 microdeletions. They share osteopoikilosis, short stature and learning disabilities as common phenotypic features. In two cases the deletion was approximately 6 megabases (Mb) in size whereas in the third patient a 3.44 Mb deletion was detected.
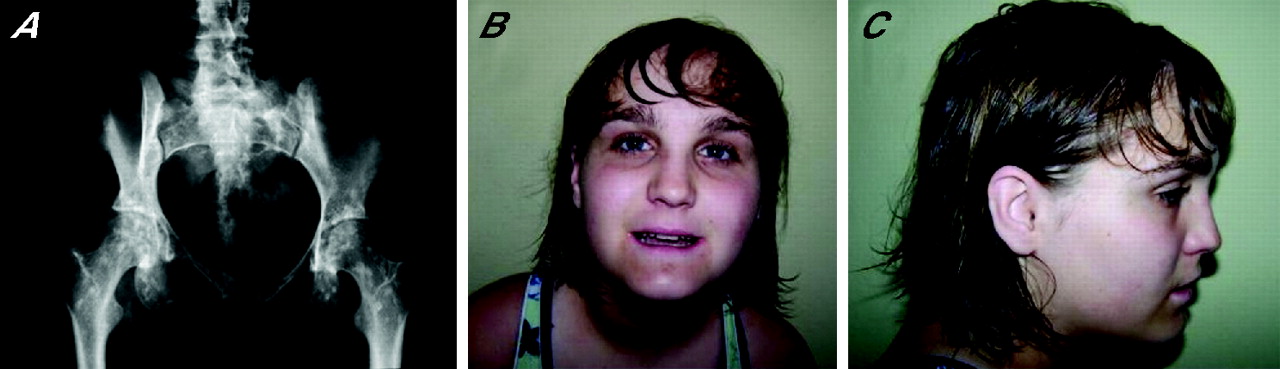
The first proband (03g1858) is a girl born at term with a birth weight of 2060 g. Pregnancy and delivery were uneventful. At the age of 6 months, she presented with failure to thrive and by the age of 1 year, length, weight and head circumference were all far below the third centile. Work-up for this failure to thrive only revealed hypertension, for which she received medication. The diagnosis of Russell-Silver syndrome was considered at that time. The girl also showed delayed neuromotor development and experienced learning difficulties requiring an individualised programme at school. Clinical evaluation at the age of 16 years revealed proportionate short stature with a weight of 31.8 kg (−4 SDs), height of 131.5 cm (−6.2 SDs), span of 131 cm and head circumference of 49 cm (−4.4 SDs). The face was mildly dysmorphic with synophrys, mild hypertelorism, broad and high nasal bridge, micrognathia and maxillary overbite. These clinical features were not reminiscent of Russell-Silver syndrome. Imaging studies revealed the presence of ectopic kidneys, and an aortogram showed on each side two renal arteries with an aberrant origin. In addition, malrotation of the small bowel, a medially positioned spleen and an unusually shaped (rectangular) liver were found. Radiographic evaluation showed multiple osteopoikilosis lesions in the pelvis, shoulders, wrists, hands and feet (fig 1A).

Clinical and radiographic features of the microdeletion syndrome. (A) Radiograph of the pelvis in patient 03g1858 showing multiple osteopoikilosis lesions in the proximal parts of the femurs and pubic bones. Facial phenotype of patient D0502619 with anteroposterior (B) and lateral (C) view (patients 03g1858 and #4818 refused clinical photographs). Parental/guardian informed consent was obtained for publication of this figure.
The second female proband (D0502619, figure 1B,C) was born at term with a weight of 2300 g. The pregnancy was complicated by oligohydramnios. The postnatal course was uneventful. Early in infancy very poor growth and development became apparent. At the age of 4 years she was diagnosed with scoliosis, type 1 Arnold-Chiari malformation and ultimately syringomelia requiring a shunt. She also had a release for a tethered spinal cord. Reflux nephropathy with small kidneys, mild hypertension and diabetes mellitus were diagnosed in childhood. In school, mild learning problems became apparent, and therefore she was put on an individualised education plan. At the age of 14 years she presented with complaints of tingling pain in the medial part of her right foot. Clinical evaluation revealed a weight of 51.3 kg (mean for age), height of 142.3 cm (−3.5 SDs) and head circumference of 53.3 cm (−0.66 SDs). Her face was round with rather deep-set eyes, bushy eyebrows and thin lips. (fig 1B,C). A thoracolumbar scoliosis was noted. The skin showed several areas of increased pigmentation. Mild swelling without other inflammatory signs was present on the right foot. The gait pattern was somewhat antalgic. Radiographic evaluation revealed numerous osteopoikilosis lesions in the distal part of the tibia and fibula as well as in the right foot. In addition, the second right metatarsal showed a thickened and irregular cortical lesion suggestive of melorheostosis at the diaphysis. At the age of nearly 16 years, she is now functioning at the level of a 10-year-old child. She is quite sociable and tends to be very articulate and repetitive.
The third patient (#4818) is the male product of the first pregnancy of an unrelated 22-year-old mother and 35-year-old father. The pregnancy was complicated by hyperemesis. There were no adverse exposures. The family history was non-contributory. Delivery was at term with weight at the third centile and length at the tenth centile. The boy failed to thrive during the first year. At age three years six months, all growth measurements were below the third centile, and development was a year delayed. Growth hormone levels and bone age were normal. At age four, he was found to have delays in fine motor skills and speech. Six secondary teeth were missing on dental radiographs. After sustaining a fracture of the right tibia, he developed clawing of the toes and electromyogram findings suggestive of sciatic nerve injury. Radiographs documented osteopoikilosis lesions within multiple bones. When seen at 12 years of age, he was noted to have mild developmental delay, with difficulties in spelling and reading. He was tiny, with all growth measurements below the third centile. His face was triangular with widely spaced eyes. There were yellowish raised areas on the skin overlying the upper chest and flank. Trichothiodystrophy was considered as a diagnosis, but his hair was normal. He was reported to have a tremor that increased with writing. The patient was seen again at 18 years of age. His final height of 152 cm and weight of 41 kg were both below the third centile. He described a slow increase in tremor, most marked upon arising or intention. His overall health had been good, and he had achieved a normal puberty. He was entering grade 12, taking applied mathematics, and having problems with English.
The microdeletion in the first proband (03g1858) was identified during the course of mapping the gene for osteopoikilosis.14 The patient showed loss of heterozygosity for a stretch of markers in the linkage interval, which resulted in a considerable reduction of the critical region and finally led (through a candidate gene approach) to the identification of LEMD3 as the causal gene for osteopoikilosis. The deletion was confirmed with fluorescence in situ hybridisation (FISH) using BAC clone RP11-305O6, as described by Van Roy et al.15 Breakpoints were further delineated using array CGH as described by Menten et al16 with a custom tiling path array for chromosomal bands 12q14–12q15. BAC clones were selected based upon the May 2004 human genome project assembly (http://genome.ucsc.edu/) (table 1, appendix).
BAC clones used for breakpoint detection in patients 1 and 2, with their name, sanger name, chromosome, start position and end position according to the NCBI 36 genome assembly
After publication of the first patient, a second patient with a similar phenotype was identified. FISH with BAC clones RP11-305O6 and RP11-361O1 confirmed the presence of a microdeletion encompassing LEMD3 on chromosome 12. As in the first proband, the size of the deletion and the position of the breakpoints were determined by array CGH using a custom tiling path array with overlapping BAC clones (fig 2). The size of the deletion in the first two cases was similar (about 6 Mb) with an overlap of about 5.3 Mb. The genomic position of the deletion in patient 1 was slightly more telomeric than that in patient 2. Karyotyping and FISH of the parents of patients 1 and 2 yielded normal results, indicating that both deletions occurred de novo.

{kind=link}
{kind=link}
Chromosomal map of the deleted region with breakpoint flanking BAC clones for patients 1 and 2, breakpoint flanking SNP markers for patient 3, RefSeq genes (UCSC Genome Browser on Human Mar. 2006 Assembly) and segmental duplications. Karyotypes are written as: 46,XX,del(12)(q14.2q15).ish del(12)(q14.3)(RP11-305O6-) arr cgh 12q14.2q15(RP11-24L23RP11-249J13)x1 de novo for patient 1, 46,XX,del(12)(q14.1q15).ish del(12)(q14.3)(RP11-305O6-,RP11-361O1-) arr cgh 12q14.1q15(RP11-467D14 RP11-444B24)x1 de novo for patient 2 and 46,XX,del(12)(q14.2q15). arr cgh 12q14.2q15(rs10506536 rs10492198)x1 de novo for patient 3.
The microdeletion in the third patient (#4818) was identified during a study of 100 children with idiopathic mental retardation and normal standard chromosomal analysis, using Affymetrix GeneChip® Human Mapping 100K arrays.9 Breakpoints were mapped to SNP rs10506536 (63342649 base pairs) and SNP rs10492198 (66780095 base pairs), indicating a 3.44 Mb deletion. Affymetrix Genechip® analysis of the parents yielded normal results, indicating that this deletion also occurred de novo. The deletion in the child was confirmed by FISH using BAC RP11-91K23. This deletion is smaller but lies entirely within the 5.3 Mb region that was deleted in both patients 1 and 2 (fig 2).
Segmental duplications or low copy repeats have been shown to play an important role in the formation of recurrent microdeletion syndromes by non-allelic homologous recombination.17–20 However, no evidence of segmental duplications or low-copy repeats was found near any of the six breakpoints in these three patients. Microdeletions may also occur without involvement of low copy repeats.12,21–23 Recent reviews estimate that only 25–50% of copy-number variants are associated with segmental duplications.24 Recurrent microdeletions that are not associated with low-copy repeats usually have different breakpoints in each case. They most likely result from breakage with subsequent nonhomologous endjoining. Given the apparent absence of low copy repeats near the 12q breakpoints described here, we suspect that a mechanism of nonhomologous endjoining may be responsible for occurrence of the microdeletions in our three cases.
In 1995, Jurenka and Van Allen reported a patient with mental retardation, short stature and a mixed sclerosing bone dysplasia reminiscent of melorheostosis.25 We suspect that this patient may have the same microdeletion as found in our three probands. Unfortunately, no DNA from this patient was available to test this hypothesis.
The similar phenotype in our three probands is remarkable. All three patients had a low birth weight and presented in infancy with failure to thrive. They subsequently showed delayed neuromotor development and finally mild mental retardation. They do not show a remarkable facial dysmorphism but all have a proportionate short stature with osteopoikilosis lesions on skeletal radiographs. One patient (#D0502619) developed a melorheostosis lesion in the foot. We have shown in previous studies that loss-of-function mutations in the LEMD3 gene can result in osteopoikilosis and/or melorheostosis lesions.14,26 However, failure to thrive, short stature and mental retardation are not observed in patients with either osteopoikilosis or melorheostosis. These findings must therefore be the result of haploinsufficiency for other contiguous genes in the microdeletion interval. In the common deleted region two interesting genes (HMGA2 and GRIP1) reside, which may account for these additional clinical problems in our patients (fig 2).
HMGA2 codes for an architectural factor belonging to the high-mobility group (HMG) of proteins. It is characterised by three conserved DNA-binding domains, AT hooks and an acidic C-terminal tail. This gene product is involved in DNA packaging and plays an important role as a transcription factor in gene regulation.27 Recently, HMGA2 was described as the putative causal gene in a patient with overgrowth, lipomas and a de novo pericentric inversion of chromosome 12.28 Battista et al reported a murine model with a constitutively expressed truncated form of Hmga2, which led to gigantism associated with lipomatosis. The authors proposed that disruption of the Hmga2 gene led to upregulated expression.29 Disruptions and rearrangements of HGMA2 leading to aberrant gene expression are a frequent observation in lipomas and other benign mesenchymal tumours.30–37 Overexpression has also been reported in malignant tumours.38 Interestingly, Zhou et al. reported a “pygmy” phenotype in Hmga2−/− murine models,39 with heterozygous mice displaying a milder phenotype (80% of the weight of wild-type mice).40 Taken together, these data are consistent with an important role for HMGA2 in growth. Hence, haploinsufficiency of this gene may result in short stature as observed in our patients. To test this hypothesis, a series of patients with idiopathic proportionate short stature will be tested for loss-of-function mutations in the HMGA2 gene.
Glutamate receptor interacting protein 1 (GRIP1) is a good candidate gene for mental retardation. GRIP1 is highly expressed in adult human and fetal brain as well as in other organ systems. The gene produces three different transcripts by alternative splicing and contains seven highly conserved domains. All GRIP1 products contain the PDZ domain, which is important in synaptic function.41GRIP1 proteins localise through their PDZ domains to α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors in cultured rat hippocampal neurons.42,43 These AMPA receptors mediate synaptic transmission through glutamate, the major excitatory neurotransmitter in the central nervous system. GRIP1 is implicated in targeting AMPA receptors to the synapse.42,44,45GRIP1 is also involved in the induction of long-term potentiation in rat hippocampal mossy fibres.46 In addition to their role in glutamergic synaptic transmission, GRIP1 products also localise to GABAergic synapses in rat hippocampal cultures and intact rat brain.47,48 Homozygous Grip1 knockout mice die as embryos.49 The heterozygous Grip1 knockout mouse phenotype has not yet been reported. The observations that GRIP1 codes for a non-redundant protein that it is highly expressed in fetal and adult human brain, and involved in glutamergic synaptic transmission, support the possibility that GRIP1 haploinsufficiency caused the learning problems in our patients.
We conclude that we have identified a newly recognisable microdeletion syndrome involving chromosome 12q14. The clinical phenotype is mainly characterised by mild mental retardation, low birth weight with failure to thrive in early infancy and proportionate short stature. The osteopoikilosis lesions on skeletal radiographs are the most distinguishing feature of this microdeletion syndrome that should facilitate recognition of this peculiar disorder among children with mental retardation and growth failure.
Acknowledgments
We are grateful to the patients and their families for their cooperation. We thank the Mapping Core and Map Finishing groups of the Wellcome Trust Sanger Institute for initial clone supply and verification, and Ivy Jennes for technical assistance. This work was made possible by grants G.0200.03 from the FWO and GOA-grant 12051203 from Ghent University.
Karen Buysse is a Research Assistant of the Research Foundation – Flanders (FWO – Vlaanderen). Jan Hellemans is funded by the Institute for the Promotion of Innovation by Science and Technology in Flanders. Marco Marra is a Scholar of the Michael Smith Foundation for Health Research. This study is supported, in part, by the Fund for Scientific Research, Flanders, with a grant for fundamental clinical research to GRM.
This text presents research results of the Belgian program of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister’s Office, Science Policy Programming (IUAP).
REFERENCES
Footnotes
-
Published Online First 12 January 2007
-
Parental/guardian informed consent was obtained for publication of figure 1.