Article Text

Abstract
A large family with dominantly inherited rhegmatogenous retinal detachment, premature arthropathy, and development of phalangeal epiphyseal dysplasia, resulting in brachydactyly was linked to COL2A1, the gene encoding proα1(II) collagen. Mutational analysis of the gene by exon sequencing identified a novel mutation in the C-propeptide region of the molecule. The glycine to aspartic acid change occurred in a region that is highly conserved in all fibrillar collagen molecules. The resulting phenotype does not fit easily into pre-existing subgroups of the type II collagenopathies, which includes spondyloepiphyseal dysplasia, and the Kniest, Strudwick, and Stickler dysplasias.
- COL2A1
- vitreoretinopathy
- brachydactyly
- chondrodysplasia
Statistics from Altmetric.com
The inherited disorders resulting from mutations in COL2A1, the gene for proα1(II) collagen form a spectrum of phenotypes, which are collectively known as the type II collagenopathies.1 These range from the lethal or perinatally lethal disorders achondrogenesis II and hypochondrogenesis,2,3 through disproportionate dwarfisms, which include spondyloepiphyseal dysplasia congenita (SEDC), the Strudwick type of spondyloepimetaphyseal dysplasia (SEMD), and Kniest dysplasia.4–9 At the milder end of the spectrum of type II collagenopathies is Stickler syndrome or arthro-ophthalmopathy.10 Other disorders include familial osteoarthritis11 and a predominantly ocular form of Stickler syndrome.12
Nine different genes code for the human fibrillar collagen molecules,13 which comprise types I, II, III, V, and XI, and all are synthesised as propeptides. The carboxyl propeptide region of each molecule is required for α chain association, which then proceed to zip up from the C to N terminus to form either homo- or heterocollagen triple helices.14 After secretion from the cell, the C-propeptides are enzymatically removed. Here we describe a large family with a COL2A1 mutation within the C-propeptide of type II collagen. The phenotype that results from this mutation does not easily fit into any of the existing categories of type II collagenopathies.
SUBJECTS, METHODS, AND RESULTS
Patients in the extended family were identified following independent referral of two family members to the genetics department of the University Hospital of Wales. Written consent to examination and DNA analysis was obtained in all cases as part of their clinical management. Affected subjects ranged in age from 22 to 60 years and presented with both ophthalmic and skeletal phenotypes. The vitreoretinal changes were characteristic with extensive lattice retinopathy, covering approximately two to three clock hours in each quadrant of some patients. Although the vitreous did not exhibit the congenital membraneous anomaly characteristic of Stickler syndrome type 1,15–17 the architecture was strikingly abnormal, with absence of the usual lamellar array. Affected subjects had a spherical mean refractive error of –1.46 dioptres (SD 1.5), which was not significantly greater than that in unaffected subjects (mean refractive error –0.71, SD 0.99, p=0.13, Mann-Whitney test). The axial length was slightly greater in affected eyes (mean 24.6 mm, SD 0.73) compared with unaffected eyes (mean 23.8 mm, SD 1.1, p=0.008, t test). A single affected subject, whose axial length had increased to 33.5 mm, following retinal detachment surgery, distorted any apparent variation in myopia between affected and unaffected subjects and is not included in the analysis of ammetropia or axial lengths. In one case (III.8) bilateral retinal detachment occurred at 13 years of age resulting in blindness. All affected subjects are at risk of rhegmatogenous retinal detachment (retinal detachment secondary to holes or tears in the retina). This is common in SEDC, Kniest dysplasia, SEMD, and Stickler syndrome.
All were considered to be of normal stature, so not all were formally measured. Of those that were, the shortest was 1.55 m (female) and tallest 1.8 m (male); there was no disproportionate stature. The skeletal changes were present in varying degrees in all but one of the affected subjects with retinopathy. The most common feature was changes to their hands. As all affected subjects were adults, it was difficult to assess the development of this feature. It was reported by the patients that at around the time of puberty they had pain and swelling of the joints in their hands, followed by disturbances in the growth of the fingers. However, the feet were normal in all cases. In the most severely affected, the fingers were recalled as being short from early childhood. The changes in the hands were highly variable with one affected subject (III.8) having a normal clinical and radiological examination (aged 34). Another (III.4) had only mild shortening of the terminal phalanges. The majority had shortening of all phalanges (figs 1 and 2), while in severe instances, involvement of the metacarpal and carpal bones was apparent together with the distal epiphyses of the radius and ulna (fig 2C). Epiphyseal changes in the hips were also variable. At its most severe, premature osteoarthritis requiring the need for hip replacement surgery was evident and occurred in III.4 at 22 years of age. In this person, there was no radiological evidence of a slipped upper femoral epiphysis, or of avascular necrosis, and the shape of the proximal femurs and bony pelvis was normal (fig 3A). Radiographs of the spine from two older subjects (fig 3B-D), who had both undergone hip replacement, were essentially normal with only minor degenerative changes compatible with age seen in one case (fig 3D). The facies appeared normal without the midfacial hypoplasia usually associated with Stickler syndrome and the more severe type II collagenopathies. None of the affected subjects had a cleft palate.
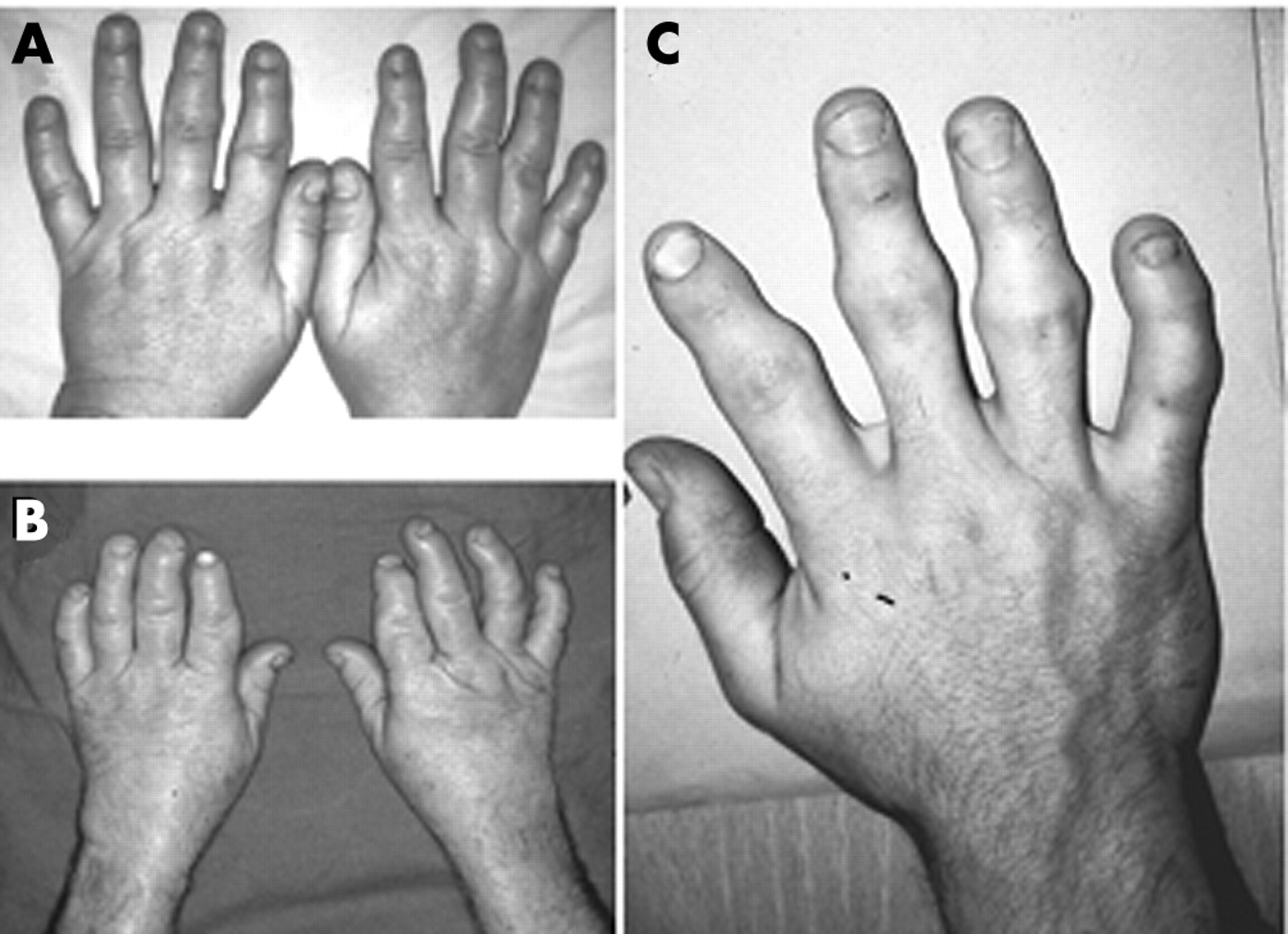
Brachydactyly in affected subjects. The hands of affected subjects II.9 (A) aged 48 years, II.2 (B) aged 55, and III.9 (C) aged 29 are shown. See fig 4 for pedigree information.

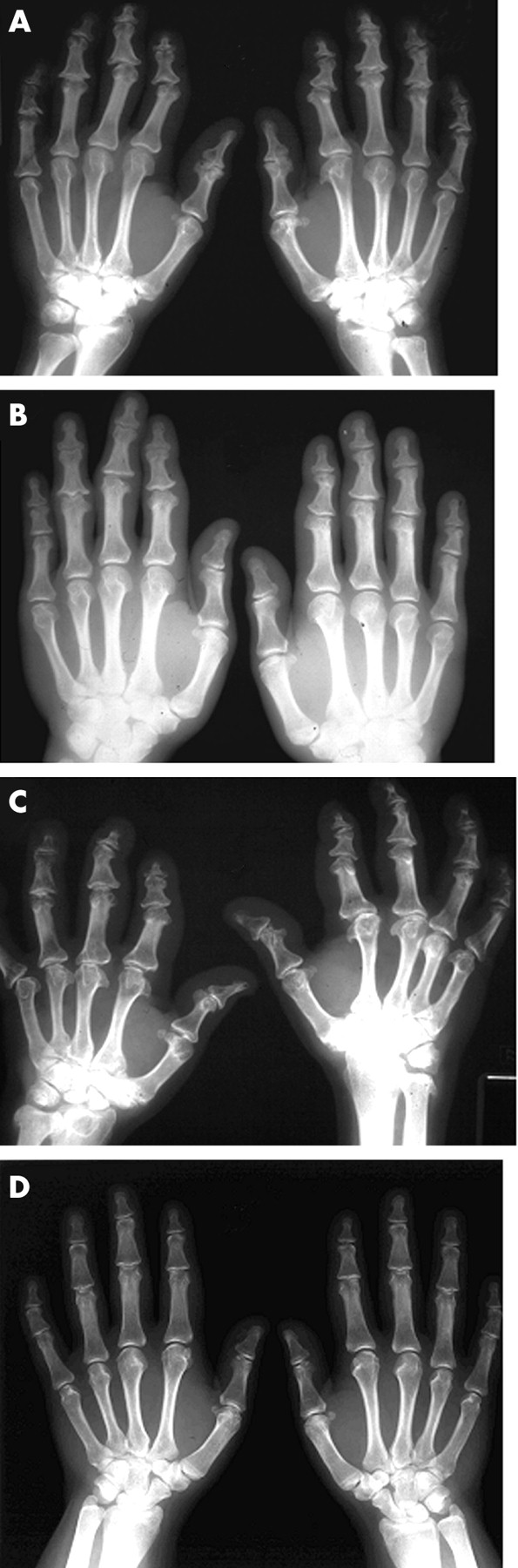
Radiographs of affected hands. Radiographs of the hands of affected subjects II.7 (A) aged 52, III.10 (B) aged 34, II.2 (C) aged 55, and III.4 (D) aged 22 are shown. See fig 4 for pedigree information. The pattern is of an epiphyseal dysplasia predominantly affecting the phalanges but in one subject (C) involvement of the carpus and distal radius is also seen. Involvement in (D) is particularly mild.
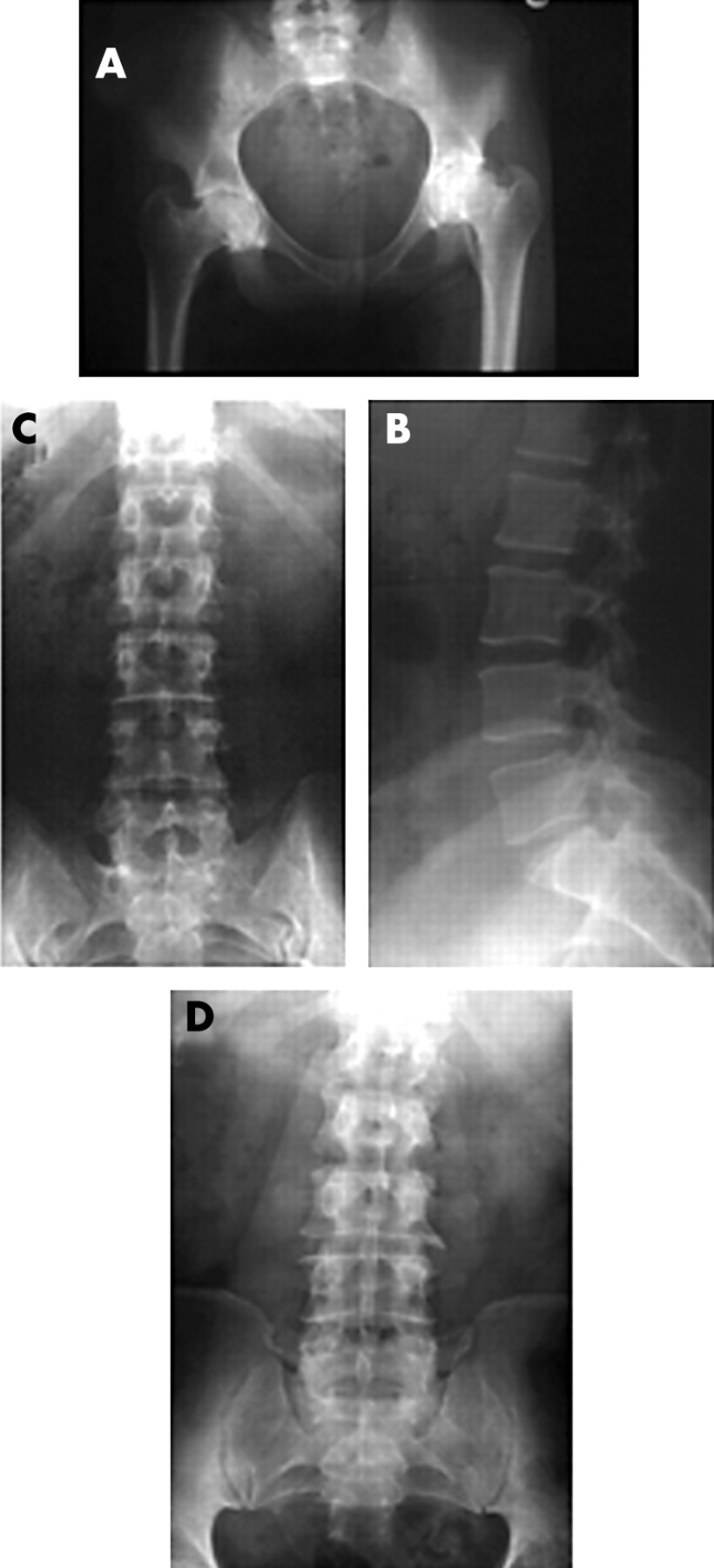
Radiographs of hips and spine. Radiographs of pelvis of III.4 (A) aged 21 before hip replacement. The lumbar spine of II.9 (B, C) aged 53 and II.2 (D) aged 60. Severe premature osteoarthritis of the hips (A) is seen but the spines are essentially normal with age related changes (D) but no evidence of dysplasia.
Because of the involvement of both the skeletal and ocular systems, the gene for type II collagen, the major component of both cartilage and vitreous, was considered to be a good candidate for the mutant locus. Polymorphic dinucleotide repeat markers on either side of the COL2A1 gene were analysed by amplification using fluorescently labelled primers and electrophoresis though a polyacrylamide gel using an ABI 377 machine and the Genescan Analysis software. Primer sequences for markers D12S85, D12S1701, D12S1661, and D12S361 were obtained from the Genethon Linkage Map.18 Lod scores were calculated using the LIPED program.19 Positive lod scores were obtained for markers on either side of COL2A1, the highest being 5.4 at zero recombination with marker D12S361.
The COL2A1 gene was analysed for mutations by using amplified DNA to sequence all 54 exons as previously described.12 A heterozygous single base (G-A) change was seen in exon 52, which converted a codon for glycine (G1105D) to aspartic acid (fig 4). In our experience this change was novel and so, using DNA from family members, its segregation with the disease was investigated. The single base change removed a BslI restriction enzyme site and this was used to test the family (fig 5). All affected family members were heterozygous for the change, which was absent in all unaffected subjects. Further tests on 50 unrelated controls (100 chromosomes) were negative for the change. These were of the same ethnicity as the affected family (white); 45 were British, three were Dutch, one was Anglo-American, and another Greek. In addition, 35 samples from white British subjects with Stickler syndrome, SEDC, or Kniest dysplasia were negative for the change as determined by sequencing. Analysis of illegitimate COL2A1 transcripts using cultured dermal fibroblasts from an affected subject showed that both alleles were amplified from the mRNA of these cells (fig 4). Amplification of transcripts was achieved essentially as previously described.20 An antisense primer in the 3` untranslated region (tggaaagtacttgggtcctttggg) was used for the reverse transcription reaction. Primers in exon 36 (ccagctggtgctaacggcgagaag) and 53 (tgaacctgctattgccctctgccc) were used in the primary PCR. Primers for the secondary PCR were nested in exons 39 (agcctggggccaagggtgagcaag) and 53 (ctggacgttggcagtgttgggagc). RT-PCR products were sequenced with a primer in exon 51 (caacaaccagattgagagcatccg). The observed heterozygosity in amplified cDNA supported the observation of a missense mutation, rather than haploinsufficiency, through nonsense mediated decay, causing the disorder.

Sequence analysis. Genomic sequencing of COL2A1 exon 52 from normal (N-G) and affected (M-G) subjects identified a heterozygous G-A single base change (arrowed). Amplification of illegitimate COL2A1 transcripts (M-C) from an affected subject showed both alleles in the mRNA sequence.

Family analysis. A region of COL2A1 containing the mutation site was amplified from individual family members, with forward primer 5`tctgtctctttcagtcaggcctgg3` and reverse primer 5`atggaagccaccattgatggtttc3`. The products were ethanol precipitated, resuspended in water, and aliquots incubated with the restriction enzyme BslI at 55°C for 16 hours. The resulting DNA molecules (arrowed) were analysed by electrophoresis in a 6% polyacrylamide gel, stained with ethidium bromide, and visualised under UV light. Samples correspond to family members shown in the pedigree above each lane. Untested subjects are enclosed with a dashed line.
Amino acid sequences for the fibrillar collagens were determined from the published cDNA sequences and aligned using the computer program PILEUP.21 By comparing the sequences for all nine human fibrillar collagens, it was seen that the altered amino acid was within a highly conserved region of the C-propeptides (fig 6). The usual convention for numbering amino acids in collagen molecules is to assign the first glycine of the triple helical domain as 1. Using this system the mutation is G1105D. However, different numbering systems have been used when describing alterations to propeptide regions. For instance, Unger et al22 numbered amino acids from the initiating methionine, but excluded amino acids encoded by exon 2, which is not expressed by mature chondrocytes. Exon assignment in some published reports is also confusing. Some reports number the 54 exons 1-54,12,20,23 while others24,25 use the numbering system of Chu and Prockop,26 where exons 1-5 are numbered 1, 1A, 2, 3, and 3B. Consequently exons 6-54 are numbered 4-52. For clarity, the different missense mutations in the C-propeptide of type II collagen are listed in table 1, amino acids are numbered from the start of the C-propeptide (DQAAG), and exons numbered 1-54.
Mutations in the C-propeptide of type II collagen
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
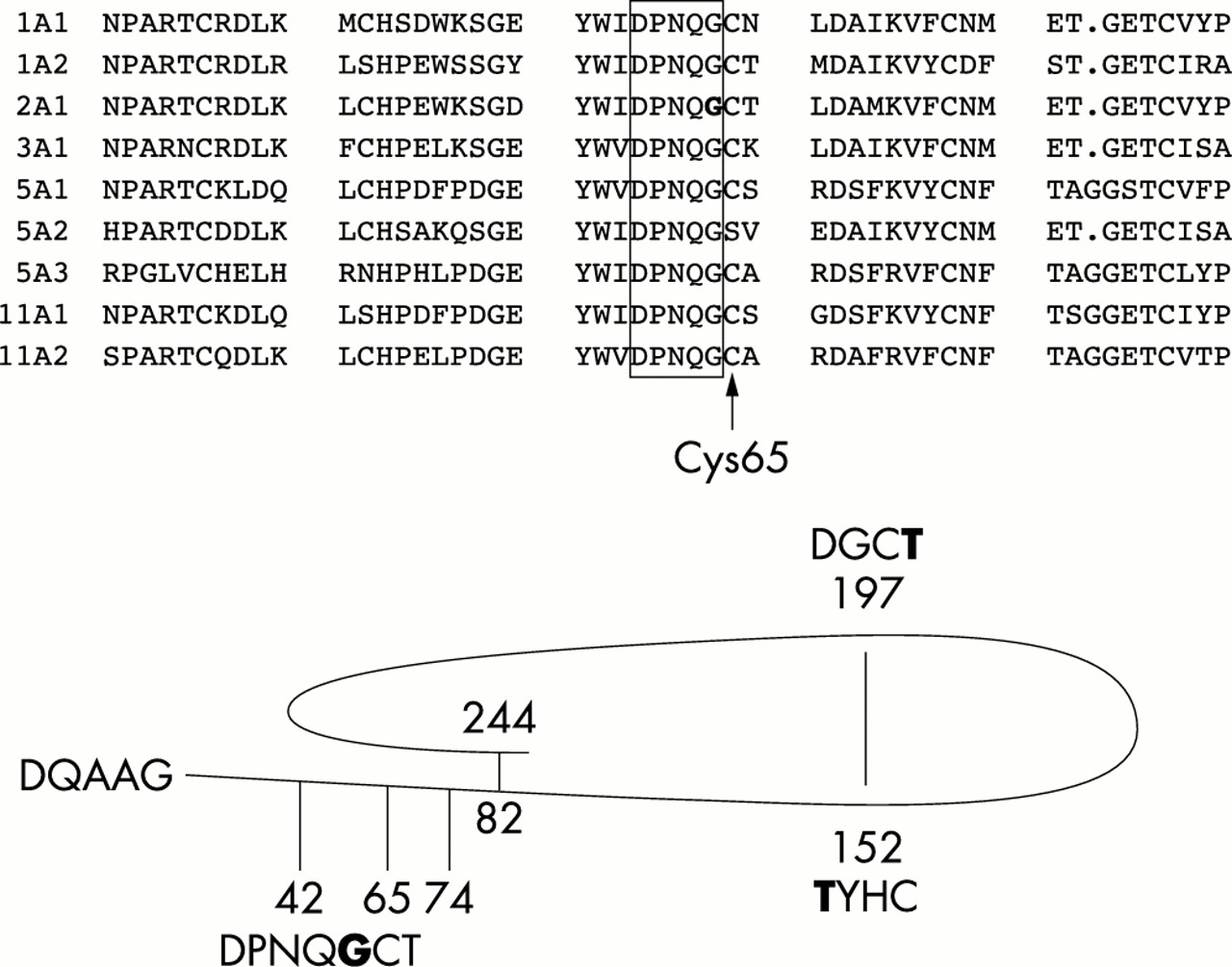
Multiple protein sequence alignment. Alignment of a region from the C-propeptides of the fibrillar collagens (as indicated), containing the type II collagen amino acid substitution. A region of five highly conserved amino acids is boxed. The mutated C-propeptide α1(II) Gly64 amino acid is in bold type. A representation of the C-propeptide indicating the position of missense mutations listed in table 1 is also shown. Again the mutated amino acids are in bold type. Amino acids are numbered from the start of the C-propeptide (DQAAG), intrachain disulphide bonds at positions 82, 152, 197, and 244 are indicated along with cysteines involved in interchain disulphide bonds at positions 42, 65, and 74.
DISCUSSION
The family we have described here have an unusual phenotype with involvement of the hands. As they have normal stature they cannot be classified as SEDC, SEMD, or Kniest dysplasia, yet they are not typical of Stickler syndrome, the most common type II collagenopathy, where stature is normal. Like Stickler syndrome this family presented with dominantly inherited retinal detachment, lattice retinopathy, and premature osteoarthritis. Stickler syndrome is highly variable, and it is not unusual for an affected subject to present with minimal features of the disorder. Indeed some mutations result in a predominantly ocular form of Stickler syndrome.12 It is, however, unusual for all affected members of a large family, with both retinal detachment and premature arthropathy, as described here, to have no instance of high myopia, cleft palate, or midfacial hypoplasia. Cleft palate, for instance, is commonly associated with mutations in COL2A1.27,28 The family described here also lacked the congenital membraneous vitreous anomaly usually present in cases of Stickler syndrome,15–17 instead exhibiting highly disorganised vitreous lamellae. This presumably reflected the different nature of the mutation which if expressed would result in mutant proα1(II) collagen chains. Unfortunately, we did not have cartilage tissue available to confirm expression of mutant protein. However, like other examples of missense mutations in COL2A1, heterozygosity of the mutation was observed in illegitimate transcripts,20 whereas premature termination mutations exhibit nonsense mediated decay20,29,30 and a homozygous normal cDNA sequence.
The skeletal phenotype varied, but the most striking trait in this family was the phalangeal epiphyseal dysplasia in the hands, which in most cases the patients reported appeared in late childhood. Brachydactyly has been noted in other cases of COL2A1 mutations. A family with an Arg704Cys mutation was described as “Stickler-like”.31 They had short stubby fingers along with myopia, a flattened midface, and mild generalised epiphyseal dysplasia. A 5 bp duplication in exon 53 resulted in a premature termination codon in the same exon and a phenotype described as spondyloperipheral dysplasia.24 In this case, where there was short stature and myopia, both hands and feet were affected with shortened digits. Again the family described here is different from these two cases.
Two other COL2A1 missense mutations have previously been described in the C-propeptide. A threonine to asparagine at position 149 resulted in hypochondrogenesis,25 while a threonine to methionine at position 198 caused SEDC.22 Like the mutation described here, these were both close to cysteines (fig 6). Certain cysteines, in the C-propeptides of type I and type III collagens, form intrachain disulphide bonds before chain association.32,33 In type II collagen the corresponding cysteines are at positions 82-244 and 152-197. Other cysteines form interchain disulphide bonds after chain association. So, unlike the mutation described here which is adjacent to Cys65 that forms an interchain disulphide bond, T149N and T198M are close to cysteines that form a single intrachain disulphide bond. It is tempting to speculate that these three mutations are disrupting disulphide bond formation, possibly delaying assembly and resulting in excess post-translational modification. Type II collagen from the patient with a T149N mutation was overmodified25; however, further work is required to elucidate the mechanism of pathogenesis in all these cases. In particular, we have not yet determined if mutant G64D α chains are capable of assembly and if mutant molecules are modified and secreted normally.
It should also be remembered that the COL2A1 gene also encodes proα3(XI) collagen34 and so these mutations will affect type XI collagen as well. Mutations in the C-propeptide region of type I collagen have also been described, in particular a mutation which substituted a cysteine in the C-propeptide region of proα1(I) collagen, disrupted an intrachain disulphide bond, and resulted in a mild form of osteogenesis imperfecta.35
In summary, the disorder in the family described here was linked to COL2A1 and a novel glycine to aspartate substitution was detected. Analysis of cDNAs suggested that both alleles were expressed, as nonsense mediated decay was not apparent. Sequencing of all 54 exons showed no other amino acid changes. Family analysis showed that the aspartate containing allele segregated with the affected subjects; this change was also absent in a panel of control chromosomes. The mutated amino acid is highly conserved in all fibrillar collagen molecules. These observations strongly indicate that this is the causative mutation in this family. Some instances of mutations in COL2A1 can result in unusual phenotypes (that is, spondyloperipheral dysplasia) that do not easily fit into the more established criteria for other type II collagenopathies, such as SEDC or the Kniest and Stickler dysplasias. This family also presented with an unusual combination of phenotypic traits. The most consistent were vitreoretinopathy (all are at risk of retinal detachment) and the developmental abnormalities that occurred in the fingers. Other aspects of the skeletal phenotype were variable; in some subjects severe premature osteoarthritis necessitated hip replacement, the youngest being at 22 years. However, there were no spinal dysplastic features. The striking developmental changes that occur to the fingers make this family highly unusual and are most likely the result of the novel nature of the mutation in the C-propeptide.
Acknowledgments
This work was funded in part by The Guide Dogs for the Blind Association, The Stanley Thomas Johnson Foundation, and The Isaac Newton Trust. We would particularly like to thank the family members for their cooperation in this study.