Article Text

Abstract
Objective: To determine the molecular basis for achromatopsia using autozygosity mapping and positional candidate gene analysis.
Design and methods: A large consanguineous Pakistani family containing six subjects with autosomal recessive complete achromatopsia was ascertained. After excluding linkage to the two known achromatopsia genes (CNGA3 and CNGB3), a genome wide linkage screen was undertaken.
Results: Significant linkage was detected to a 12 cM autozygous segment between markers D1S485 and D1S2881 on chromosome 1p13. Direct sequence analysis of the candidate gene GNAT2 located within this interval identified a frameshift mutation in exon 7 (c842_843insTCAG; M280fsX291) that segregated with the disease.
Conclusions: The GNAT2 gene codes for cone α-transducin, the G protein that couples the cone pigments to cGMP-phosphodiesterase in phototransduction. Although cone α-transducin has a fundamental role in cone phototransduction, mutations in GNAT2 have not been described previously. Since mutations in the CNGA3 gene may cause a variety of retinal dystrophies (complete and incomplete achromatopsia and progressive cone dystrophy), GNAT2 mutations may also prove to be implicated in other forms of retinal dystrophy with cone dysfunction.
- achromatopsia
- α-transducin
- GNAT2
- autozygosity mapping
Statistics from Altmetric.com
The photoreceptor dystrophies are an important cause of childhood blindness and represent a broad spectrum of diseases. Cone and cone-rod dystrophies are characterised by early involvement of the cone receptors and usually result in profound visual loss with abnormal colour vision and sensitivity to light. Various subtypes have been identified on the basis of natural history, psychophysical, and electrophysiological testing, a notable distinction being between stationary and progressive types.1,2 Cone and cone-rod dystrophies are genetically heterogeneous and may be inherited as autosomal dominant, recessive, or X linked traits. There is marked clinical and locus heterogeneity even within these subgroups. Furthermore, mutations in a single gene may cause a variety of phenotypes (Retnet database, http://www.sph.uth.tmc.edu/Retnet/home.htm).
Complete achromatopsia or rod monochromatism (MIM 216900, 262300, 603096) is a stationary cone dystrophy with an incidence of ∼1 in 30 000.3,4 The condition is characterised by an absence of functional cone photoreceptors in the retina. Affected subjects usually present in infancy with nystagmus, poor visual acuity (20/200-20/400), photophobia, and complete colour blindness. Fundal examination is normal, but electroretinography shows absent photopic (light adapted or cone) responses and normal scotopic (rod) responses. Subjects with incomplete achromatopsia retain some colour vision and have better visual acuity.2
Achromatopsia is recessively inherited and genetically heterogeneous. To date, two achromatopsia genes, CNGA35–7 and CNGB3,8–10 have been characterised. CNGA3 and CNGB3 code for the alpha and beta subunits of the cGMP gated channel in cone cells, respectively. Germline CNGA3 mutations have been detected in ∼20% of achromatopsia kindreds and although CNGB3 mutations are thought to account for more cases, it is likely that there is further genetic heterogeneity.7 A third achromatopsia locus (ACHM1) on chromosome 14 was suggested by the report of a patient with achromatopsia and isodisomy for chromosome 14.11 In addition to causing complete achromatopsia, CNGA3 mutations may also be associated with incomplete achromatopsia or severe progressive cone dystrophy.7
Autozygosity mapping in consanguineous families is a powerful strategy for localising recessive genes even in the presence of locus heterogeneity.12–17 To elucidate the molecular basis of achromatopsia further, we investigated a large consanguineous family originating from the Indian subcontinent. After excluding linkage to CNGA3 and CNGB3, we performed a genome wide scan using an autozygosity mapping strategy, localised a novel achromatopsia locus (ACHM4) at 1p13, and identified a germline mutation in a candidate gene, GNAT2, that segregated with the disease.
PATIENTS AND METHODS
Patients
The pedigree of the three generation consanguineous Pakistani family containing six subjects with achromatopsia (and 10 unaffected relatives) is shown in fig 1. All patients had a history of nystagmus from infancy, marked photophobia, defective colour vision, and poor visual acuity. Fundus examination showed mild foveal atrophic changes. The visual impairment was non-progressive. One affected subject had electroretinography which showed normal rod responses but absent cone function, consistent with a diagnosis of achromatopsia. DNA was available as indicated (numbered subjects). Informed consent was obtained from the participants and the study was approved by the relevant local research ethics committees.

Family pedigree. Solid symbols indicate clinically affected subjects and open symbols represent unaffected subjects. DNA was available from subjects 1-16. Haplotype results for selected subjects are also illustrated.
Molecular genetic studies
DNA was isolated from blood samples by standard techniques.18 Linkage to CNGA3 and CNGB3 was excluded by typing microsatellite markers flanking each gene (CNGA3: D2S133, D2S2175, D2S2311, D2S2187, and CNGB3: D8S167, D8S1119, D8S467). In addition, mutation analysis of CNGA3 and CNGB3 was performed by direct sequencing of exons and flanking intronic sequences (primer sequences are available on request). A 10 cM genome wide linkage screen was then undertaken using fluorescently labelled microsatellite markers from the Research Genetics version 10 mapping panel. PCR amplifications were performed in 10 μl reactions with 20 ng of genomic DNA using standard conditions and “Thermoprime plus” Taq polymerase. The PCR products were then pooled into panels, diluted, and analysed on an ABI 377 DNA analyser. PCR product sizes were determined by reference to an internal standard (TAMRA GeneScan-500 size standard) using Genescan v 3.1.2 and Genotyper v 2.5.2 software (Applied Biosystems Ltd, Warrington, UK).
Mutation analysis of candidate gene
GNAT2 mutation analysis was undertaken with primers described by Magovcevic et al19 (table 1). Each of the eight coding exons (and the exon-intron boundaries) were amplified separately. PCR products from both affected and unaffected subjects were sequenced with a BigDye (version 3) Terminator Cycle Sequencing Kit on both the forward and reverse strands using an ABI 377 DNA analyser (Applied Biosystems). The mutation was numbered according to the nucleotide sequence GenBank Accession number Z18859.
Primers for PCR amplification of exons in the human cone α-transducin gene, GNAT222
Statistical methods
Two point lod scores were calculated using the MLINK program of the LINKAGE (version 5.1) package20 (http://www.hgmp.mrc.ac.uk), assuming a fully penetrant autosomal recessive gene with a disease allele frequency of 0.001. Alleles for the marker loci were assumed to be codominant and to occur at equal frequencies, because population allele frequencies were not available.
RESULTS
Analysis of CNGA3 and CNGB3
Genotyping of 16 subjects (six affected and 10 unaffected) for microsatellite markers flanking CNGA3 (D2S133, D2S2175, D2S2311, D2S2187) and CNGB3 (D8S167, D8S1119, D8S467) showed no evidence of linkage and no evidence of homozygosity by descent in affected subjects. Furthermore, direct sequencing of all the CNGA3 and CNGB3 coding exons in three affected subjects did not show a germline mutation.
Autozygosity mapping
In order to map a novel achromatopsia locus, a genome wide linkage screen was performed with DNA from four affected family members. Thus, 404 highly polymorphic tri- and tetranucleotide markers from the Research Genetics v10 marker set (which covers the whole genome at an average density of 10 cM) were analysed. Initial inspection of the results showed seven homozygous regions and the whole kindred was then typed for the markers in each of these intervals. Following genotyping of additional family members, linkage was excluded in six of the seven candidate regions, but genotyping results for a 20 cM autozygous region on chromosome 1 were consistent with linkage. Typing of an additional 10 markers (D1S495, D1S485, D1S429, D1S239, D1S2688, D1S248, D1S2651, D1S221, D1S2726, and D1S2881) narrowed the homozygous segment to a 12 cM region between markers D1S485 and D1S2881. A maximum two point lod score was obtained at GATA133A08 (Zmax=3.10 at θ=0) assuming equal allele frequencies of 0.2.
Mutation analysis of GNAT2
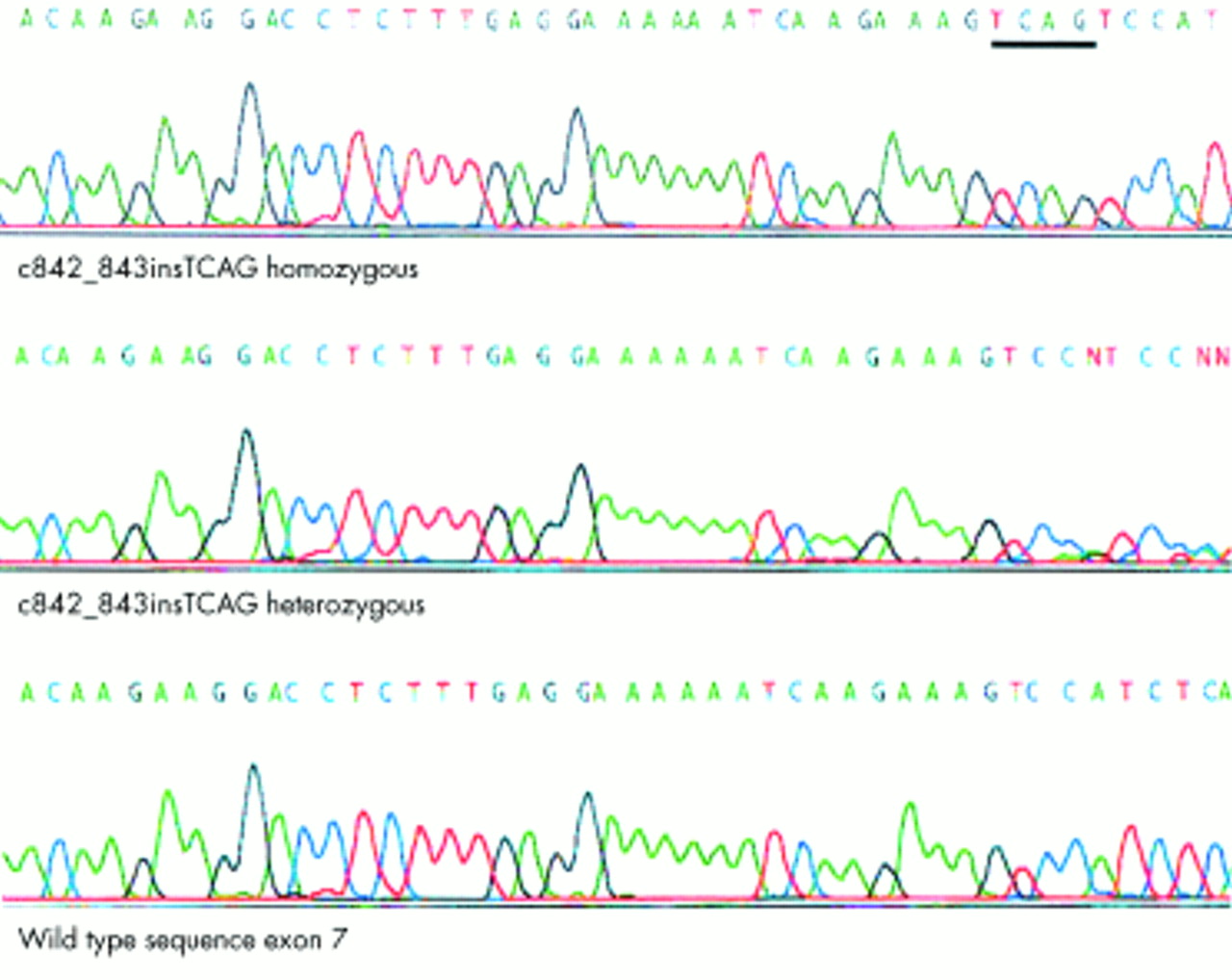
Inspection of the Human Genome Draft sequence (Ensembl at the Sanger Centre, http://www.ensembl.org and the Human Genome Browser at University of California, Santa Cruz, http://genome.ucsc.edu/) showed that the GNAT2 gene was contained within the critical interval for our novel achromatopsia locus (ACHM4). GNAT2 encodes the cone specific α-transducin protein and therefore represented an excellent candidate gene. Direct sequencing of each of the eight exons of GNAT2 showed wild type sequence, except for a 4 bp insertion (c842_843insTCAG) in exon 7 (fig 2) that results in a frameshift mutation (M280fsX291). A stop codon is present 10 codons downstream of the insertion. The mutant allele therefore encodes a mutant protein 290 amino acids in length, the first 280 residues of which are wild type sequence, with 63 residues of the wild type protein being truncated at the carboxy-terminal (fig 3). All affected subjects within the family were homozygous for the c842_843insTCAG mutation, all obligate carriers were heterozygous, and unaffected at risk subjects were heterozygotes or homozygous wild type.
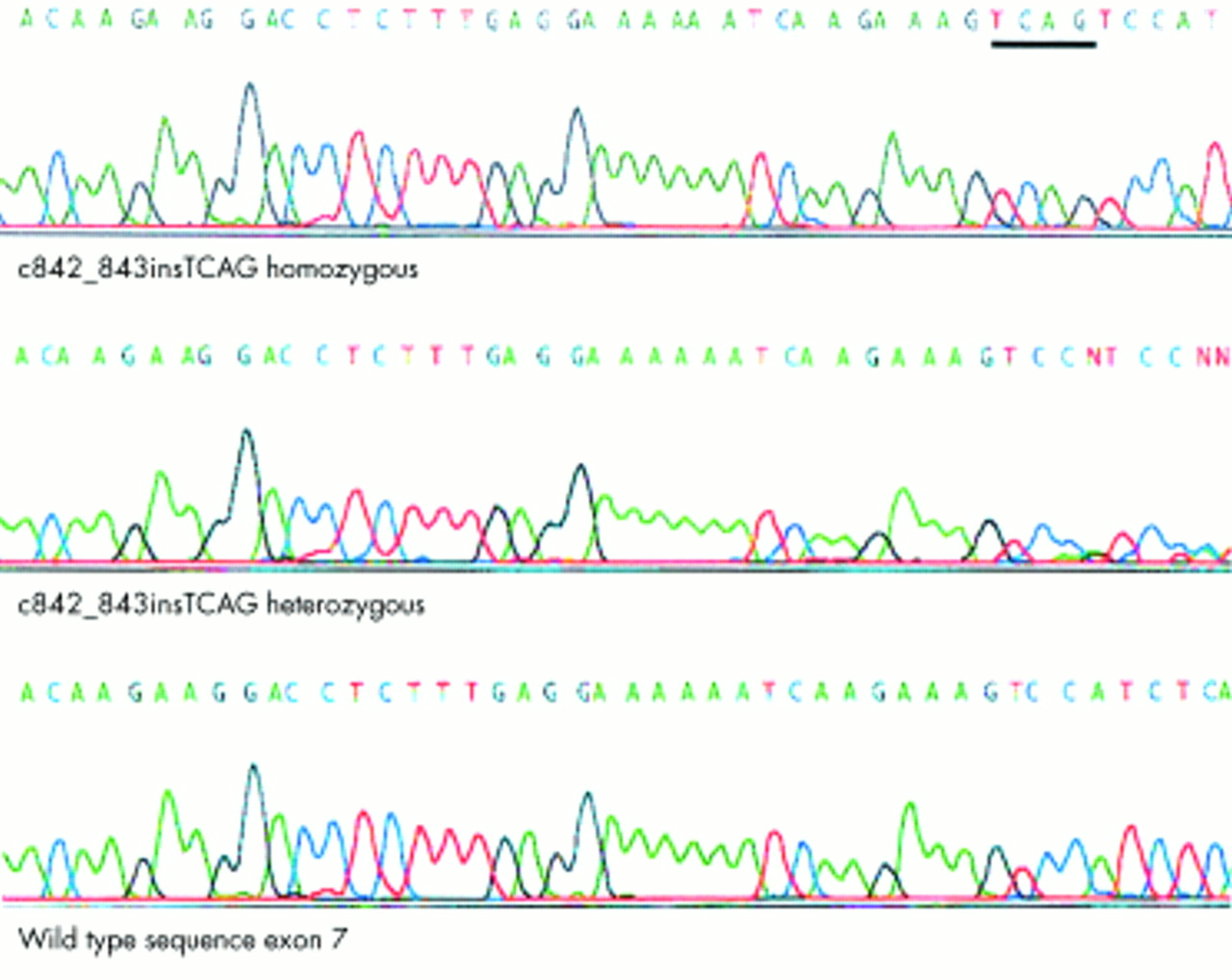
c842_843insTCAG GNAT2 mutation in achromatopsia: electropherograms for a homozygote, heterozygote, and wild type sequences.
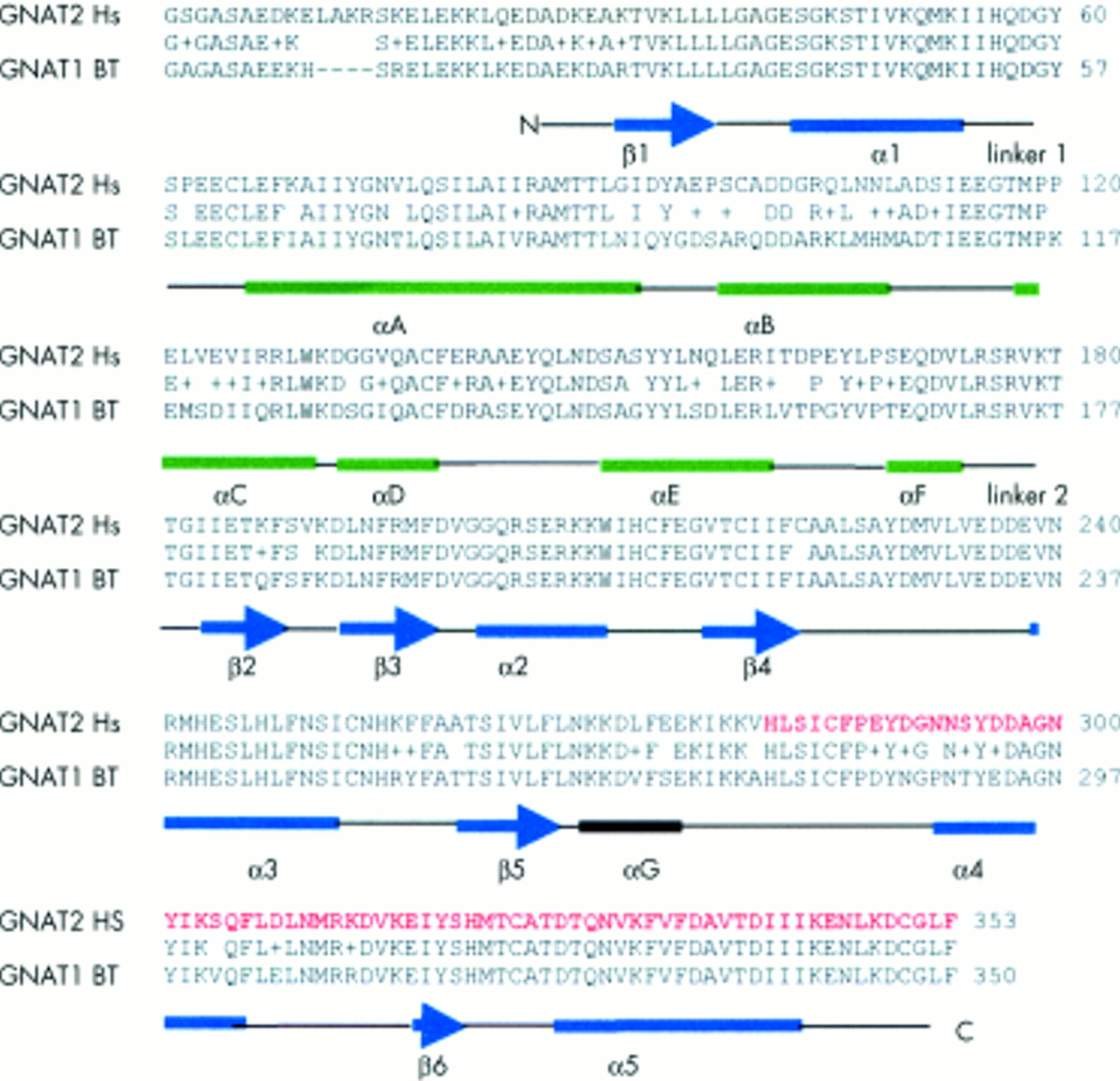
Comparison of amino acid sequences of bovine rod α-transducin (GNAT1 BT) and human cone α-transducin (GNAT2 Hs) showing 81% homology. Amino acids predicted to be absent from the mutant protein are shown in red. Structural domains as described by Lambright et al32 are indicated. Two major functional domains have been defined (a) GTPase domain (in blue), which is common to the members of the GTPase family, consisting of five helices (α1-α5) surrounding a six stranded β sheet (β1-β6) and (b) an α-helical domain (in green) consisting of one long central αA helix surrounded by five shorter helices (αB-αF). These domains are linked by two extended linker strands (1 and 2). Between the 2 domains lies a deep cleft where the nucleotide is bound. Diagram derived from a blastp search (http://www.ncbi.nih.gov/BLAST/) of bovine α-transducin-gi 121031 and human α-transducin-gi 232151.
DISCUSSION
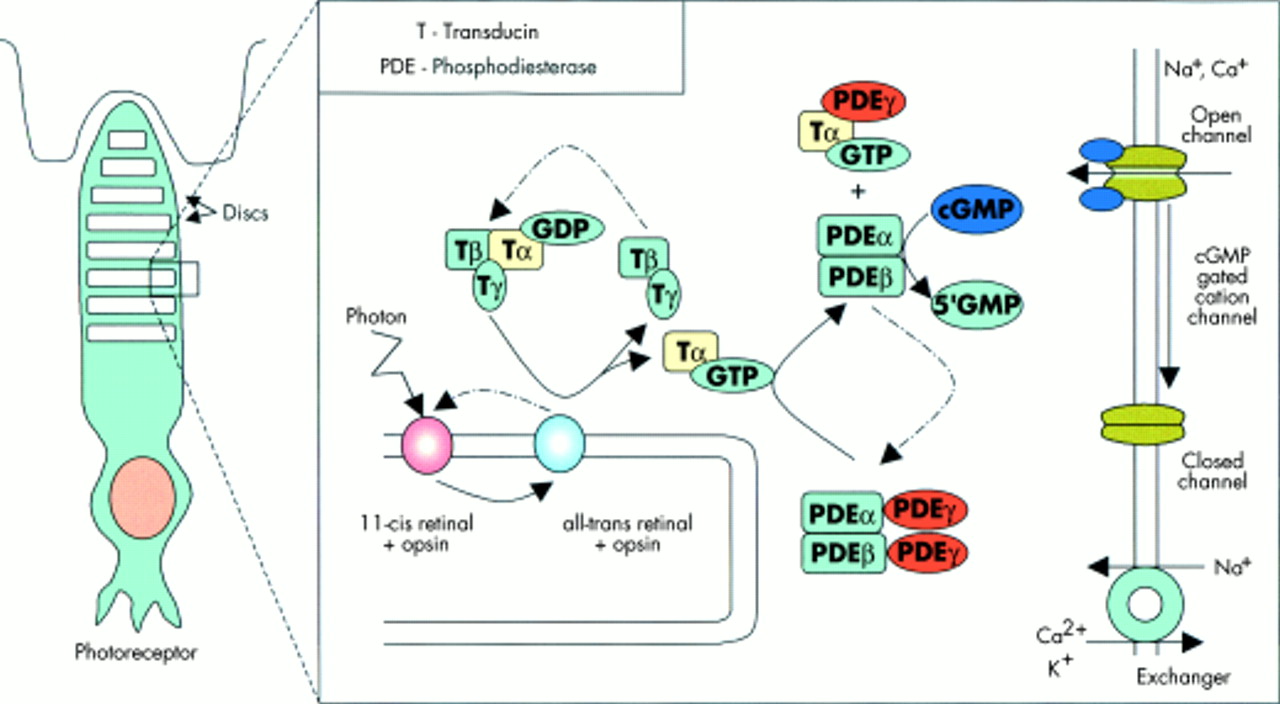
We have established homozygous GNAT2 mutations as a novel cause for achromatopsia. CNGA3 and CNGB3 encode the alpha and beta subunits of the cone photoreceptor cGMP-gated channel, which is a critical component of the cone phototransduction cascade.5–10 In cone photoreceptors, light activated photopigment interacts with transducin, a three subunit guanine nucleotide binding protein (G protein), stimulating the exchange of bound GDP for GTP. The cone specific α-transducin subunit which is bound to GTP is then released from its β and γ subunits and activates cGMP-phosphodiesterase by removing the inhibitory γ subunits from the active site of this enzyme. cGMP-phosphodiesterase lowers the concentration of cGMP in the photoreceptor which results in the cGMP gated channel closing and hyperpolarisation of the photoreceptor21–24 (fig 4). GNAT2 encodes the cone specific α-transducin subunit.25 Thus, the finding of a germline GNAT2 mutation in achromatopsia is consistent with the known function of the GNAT2 gene product. Furthermore, mutations in human rod specific α-transducin protein, GNAT1, which is 83% homologous to cone α-transducin,25,26 cause congenital stationary night blindness, a stationary retinal dystrophy affecting the rod cells.27 The frameshift mutation in the reported family would, if translated, result in a truncated protein that lacks 63 amino acids from the carboxy-terminus. This region of GNAT2 contains important functional domains of α-transducin. Specifically, amino acid sequences 310-313 and 342-345 of α-transducin have been shown to interact with rhodopsin28 and phosphodiesterase-γ interacts with multiple sites (α3, α4, and β6 of the GTPase domain) on the carboxy-terminal of activated α-transducin29 (fig 3). Further studies are required to determine if there are any phenotypic (clinical, electrodiagnostic, or psychophysical) differences between GNAT2 associated achromatopsia and that associated with CNGA3 and CNGB3 mutations. Although the cone specific α-transducin was first described in 1986,24 no disease phenotypes have previously been associated with mutations in GNAT2. Mutation analysis of 526 patients with retinitis pigmentosa30 and 66 patients with Stargardt's disease19 were all negative. As GNAT2 transcripts have also been detected in human fetal cochlea, mutation analysis was performed in 140 Usher syndrome type I and II patients; this was also negative.31 Further analysis is required to define the contribution of GNAT2 mutations to achromatopsia in different ethnic groups. In view of the multiple phenotypes associated with CNGA3 mutations7, further analysis of GNAT2 is also indicated in incomplete achromatopsia and in other candidate diseases, such as the progressive cone dystrophies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The phototransduction pathway. In cone photoreceptors, light activated photopigment interacts with transducin, a three subunit guanine nucleotide binding protein (G protein), stimulating the exchange of bound GDP for GTP. The cone specific α-transducin subunit which is bound to GTP is then released from its β and γ subunits and activates cGMP- phosphodiesterase by removing the inhibitory γ subunits from the active site of this enzyme. cGMP-phosphodiesterase lowers the concentration of cGMP in the photoreceptor, which results in the cGMP gated channel closing and hyperpolarisation of the photoreceptor.21–24
Acknowledgments
We thank the patients and their family members who contributed to this study. We acknowledge financial support from the Wellcome Trust (RCHX08477), the British Retinitis Pigmentosa Society, the Guide Dogs for the Blind Association, and Birmingham United Hospitals Endowment Fund. We are grateful to Andrew Jackson for helpful discussions.
NOTE ADDED IN PROOF Kohl et al (Am J Hum Genet, in press) have also described GNAT2 mutations in achromatopsia.