Article Text

Abstract
LEOPARD syndrome is an autosomal dominant disorder with multiple lentigines, congenital cardiac abnormalities, ocular hypertelorism, and retardation of growth. Deafness and genital abnormalities are less frequently found. We report a father and daughter and a third, unrelated patient with LEOPARD syndrome. Recently, missense mutations in the PTPN11 gene located in 12q24 were found to cause Noonan syndrome. All three cases of LEOPARD syndrome reported here have a Y279C mutation in the PTPN11 gene. We hypothesise that some PTPN11 mutations are associated with the typical Noonan syndrome phenotype and that other mutations, such as the Y279C mutation reported here, are associated with both the Noonan syndrome phenotype and with skin pigmentation anomalies, such as multiple lentigines or café au lait spots.
- LEOPARD syndrome
- Noonan syndrome
- PTPN11 mutations
Statistics from Altmetric.com
The acronym LEOPARD syndrome was introduced by Gorlin et al1 for the combination of multiple Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormalities of the genitalia, Retardation of growth, and Deafness. Fewer than 100 cases have been reported. It is an autosomal dominant condition with variable expression for which a gene location has so far not been reported. Mental retardation, if present, is usually mild and the main features of the syndrome are multiple lentigines in combination with a congenital heart malformation. This is frequently pulmonary stenosis or subvalvular muscular aortic stenosis, which may be associated with a progressive (obstructive) cardiomyopathy2 and abnormal ECG findings.3
Noonan syndrome4 is diagnosed in about 1:1000-2500 newborns. This autosomal dominant syndrome is characterised by short stature, facial dysmorphism including hypertelorism and ptosis, a low posterior hairline and webbed neck, and a heart defect. Pulmonary stenosis or hypertrophic cardiomyopathy are the most common cardiopathies found in patients with Noonan syndrome. Mental retardation, present in 15-35% of the cases, is usually mild.5 Skin manifestations are present in children and adults with Noonan syndrome and clinical overlap with neurofibromatosis has been reported.6–8 Pigmented naevi have been reported in 25%, café au lait spots in 10%, and lentigines in 3% of cases.5 The combination of facial dysmorphism and heart defects such as pulmonary stenosis or hypertrophic cardiomyopathy is found in both Noonan and LEOPARD syndromes.
Noonan syndrome is a heterogeneous disorder.9 Some families with LEOPARD syndrome and some Noonan syndrome families with café au lait spots show clinical overlap with neurofibromatosis type I, but the phenotype did not segregate with markers from the NF1 gene in these families.7 LEOPARD syndrome has features in common with both neurofibromatosis and Noonan syndrome.
Noonan syndrome has been linked to markers on chromosome 12q24 in some families.10,11 Missense mutations in the PTPN11 gene, located in 12q24, were found to cause Noonan syndrome in 50% of the patients examined.12 We performed mutation analysis of the PTPN11 gene in three patients with a LEOPARD syndrome phenotype, a father and daughter and an unrelated case.
CASE REPORTS
Patients 1 and 2
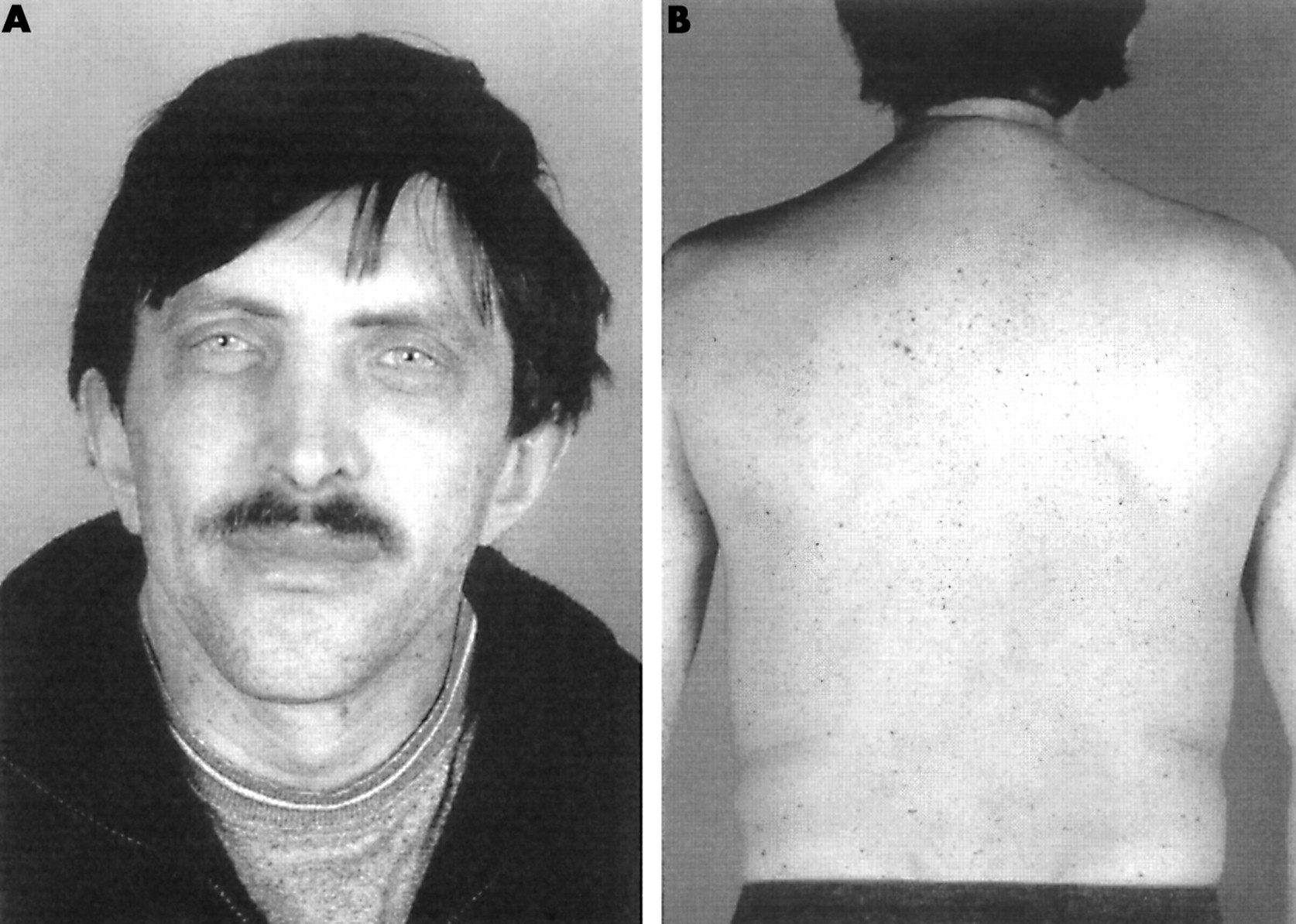
Clinical and molecular studies were performed in a father and his daughter with LEOPARD syndrome. The index patient was the daughter, who presented with short stature (height below the 3rd centile), relative macrocephaly (occipitofrontal circumference (OFC) on the 90th centile for age), and hypertrophic cardiomyopathy (asymmetrical septal hypertrophy which was stable over time). She had a relatively broad, asymmetrical chest (internipple distance 18.5 cm, 75th centile for age), hypertelorism (ICD 4.2 cm, >97th centile) with epicanthic folds, and mild mental delay. Webbing of the neck was not present. Her skin showed numerous, small, dark pigmented spots (fig 1). Similar skin features were present in the father, a healthy man with a medical history of recurrent pneumonia and constipation during childhood. During childhood he had failed to thrive and had short stature. His parents were 27 and 26 years old when he was born. He was the third in a family of six children. At that time he was the first and only person with these medical features in his family. His adult height (171.5 cm with mild kyphosis) and OFC are within normal limits (57.5 cm). His chest is broad with an internipple distance of 26 cm (>97th centile) and a pectus carinatum. He and his wife had a normal cardiac evaluation at the time when the daughter was diagnosed with the cardiomyopathy. His facial features (fig 2) were remarkable because of the very bright blue eyes which were unusual in his family. Hypertelorism was not present (ICD 3.4 cm, 50th-75th centile) and there was no webbing of the neck. His skin showed multiple, small, and very darkly pigmented spots (fig 2). His school career was normal but short and he started work at a young age. Cytogenetic studies in both the father and the daughter were normal and eye examination showed no Lisch nodules in the irides. In this family with LEOPARD syndrome, mutation and microdeletion analysis of the NF1 gene was performed but no abnormality was found.

(A) Patient 1 at the age of 12 years showing hypertelorism and epicanthic folds and (B) multiple, dark, pigmented lentigines on the trunk.

(A) Father of patient 1 (= patient 2) at the age of 41 years showing facial features and (B) multiple lentigines.
Patient 3
This 1 year old girl is one of twins. She was born at 35 weeks’ gestation and had a birth weight of 2370 g. The co-twin weighed 2630 g. The father was 41 and the mother 30 years old when the twins were born. This girl presented with a systolic heart murmur. Cardiac investigation showed hypertrophic cardiomyopathy with a mild left outflow tract obstruction (20 mm Hg), and a left anterior bundle branch block. She had facial features of Noonan syndrome with bilateral mild ptosis of the eyelids and hypertelorism. At 12 months of age, her height was on the 10th centile, weight below the 3rd centile, and head circumference on the 25th centile. The height and weight of the co-twin were on the 25th and the 10th centile, respectively. Psychomotor development was adequate. Multiple dark lentigines (>10) and three larger café au lait spots were present. Both LEOPARD syndrome and neurofibromatosis-Noonan syndrome were considered in this child. No similar phenotype was present in the parents or her co-twin. Because of the strong suspicion of neurofibromatosis, a mutation and microdeletion analysis of the NF1 gene was performed and showed normal results. Routine cytogenetic analysis with G banding on peripheral lymphocytes was normal. After blood sampling, a long clotting time was noted in this child but this was not investigated further.
Further analyses of these patients for PTPN11 mutations were performed because of the clinical overlap between Noonan syndrome, LEOPARD syndrome, and neurofibromatosis type 1.
METHODS
Primer sequences and PCR conditions are available upon request. Amplicons typically included a 50 bp intronic sequence on either side of the exon. PCR amplification of genomic DNA was followed by denaturation at 95°C for two minutes, slow cooling to 45°C for 30 minutes, and a final incubation at 45°C for 30 minutes to allow heteroduplexes to form. DHPLC screening was done using a WAVE 3500HT system (Transgenomic, Cheshire, UK). Gradients and column temperatures were used as recommended by the Wavemaker 4.1 software. Amplicons with an aberrant elution profile were subsequently sequenced with Big-dye terminator cycle sequencing (Applied Biosystems, Foster City, CA, USA) as recommended by the manufacturer and analysed on an ABI-3100 (Applied Biosystems).
RESULTS
In all three patients, the same mutation was found changing the adenine at cDNA position 836 into a guanine. This mutation in exon 7 of the PTPN11 gene (fig 3) changes the tyrosine to a cysteine codon at position 279 of the protein (Y279C). The parents of patient 3 were not available for molecular analysis. In a screening for PTPN11 mutations in 60 patients with Noonan or related syndromes, the Y279C mutation was never observed except in the cases reported here. All subjects had the same ethnic background.

{kind=link}
{kind=link}
{kind=link}
Sequencing result showing the wild type sequence of exon 7 in a control sample and the heterozygous change of an adenine at cDNA position 836 into a guanine in patient DNA.
DISCUSSION
The present reported cases indicate that, at least in some patients, LEOPARD syndrome, neurofibromatosis-Noonan syndrome, and Noonan syndrome are part of a clinical spectrum caused by mutations in the PTPN11 gene. We previously reported a de novo NF1 mutation in a patient with features of LEOPARD syndrome and a muscular outflow tract stenosis of the left cardiac ventricle.13 This subject presented with lentigines and only four café au lait spots larger than 1.5 cm, illustrating the clinical overlap between neurofibromatosis type 1 and LEOPARD syndrome. The cutaneous features of the cases reported here with a PTPN11 mutation (lentigines and small café au lait spots) could be identical to the skin features in NF1, especially in young children. Therefore, analysis of PTPN11 mutations might be helpful in these situations.
We identified the same PTPN11 mutation in two unrelated families with a LEOPARD phenotype, and we did not find this particular mutation in a larger screening for mutations in a group of 60 subjects with Noonan or a related syndrome (unpublished data).
The PTPN11 gene encodes the non-receptor protein tyrosine phosphatase SHP-2. It contains two Src homology 2 (SH2) domains and a protein tyrosine phosphatase domain (PTP). In the inactive (closed) state, the amino N-SH2 domain binds the PTP domain resulting in a steric block of the catalytic site of the PTP domain.14 When the N-SH2 domain binds a phosphopeptide the equilibrium shifts towards the “open” conformation and the steric block of the catalytic site is relieved, resulting in a catalytically active SHP-2 protein.
The Y279C mutation is localised in the PTP domain of the protein. The tyrosine residue at position 279 is conserved in mouse, rat, chicken, Drosophila, and C elegans proteins. It is also conserved in related human protein tyrosine phosphatases such as PTPN6 (PTP-1C), SHP-1, and RPTP type T.
The tyrosine residue at position 279 is involved in an intramolecular interaction of the PTP domain with the N-SH2 domain. Mutation of residue 279 most likely interferes with this interaction and results in a constitutively active PTP domain.
It is not surprising that the reported mutation is a missense mutation in an interacting portion of the N-SH2 and PTP domains because all reported PTPN11 mutations in Noonan syndrome are of this type.12 Therefore, we believe that the Y279C mutation is responsible for the LEOPARD phenotype in the reported patients.
We hypothesise that some PTPN11 mutations are associated with the typical Noonan syndrome phenotype and that other mutations, such as the Y279C mutation reported here, are associated with skin pigmentation anomalies, such as multiple lentigines or café au lait spots, in addition to the Noonan phenotype. In the light of these findings, PTPN11 mutation analysis should be carried out in a broader group of patients including those with neurofibromatosis-Noonan syndrome, families with “café au lait spots only” not linked to the NF1 locus,15,16 and adults suspected of having neurofibromatosis type 1 but without neurofibromas or Lisch nodules.
In conclusion, LEOPARD syndrome is a heterogeneous disorder. In some, a NF1 mutation is present, and in others a PTPN11 mutation. Further analysis in a larger group of LEOPARD syndrome patients might even suggest a third locus.
Acknowledgments
The authors thank the patients for their kind collaboration, Francis van der Lubbe for the clinical pictures, and L Messiaen (Medical Genetics Department, Ghent, Belgium) for sequence analysis of the NF1 gene in case 3. This research was sponsored in part by the Belgian Foundation for Research in Paediatric Cardiology. EL is a part time clinical researcher of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO-Vlaanderen).