Article Text

Abstract
EEC syndrome is an autosomal dominant disorder with the cardinal signs of ectrodactyly, ectodermal dysplasia, and orofacial clefts. EEC syndrome has been linked to chromosome 3q27 and heterozygous p63 mutations were detected in unrelated EEC families. In addition, homozygous p63 null mice exhibit craniofacial abnormalities, limb truncations, and absence of epidermal appendages, such as hair follicles and tooth primordia. In this study, we screened 39 syndromic patients, including four with EEC syndrome, five with syndromes closely related to EEC syndrome, and 30 with other syndromic orofacial clefts and/or limb anomalies. We identified heterozygous p63 mutations in three unrelated cases of EEC syndrome, two Iowa white families and one sporadic case in a Filipino boy. One family is atypical for EEC and has features consistent with Hay-Wells syndrome. In this family, the mutation ablates a splice acceptor site and, in the other two, mutations produce amino acid substitutions, R280C and R304Q, which alter conserved DNA binding sites. Germline mosaicism was detected in the founder of the mutation in one case. These three cases show significant interfamilial and intrafamilial variability in expressivity. We also screened p63 in 62 patients with non-syndromic orofacial clefts, identifying an intronic single nucleotide polymorphism but finding no evidence of mutations that would explain even a subset of non-syndromic orofacial clefts. This study supports a common role for p63 in classical EEC syndrome, both familial and sporadic, but not in other related or non-syndromic forms of orofacial clefts.
- p63
- EEC syndrome
- cleft
- mutational analysis
Statistics from Altmetric.com
Orofacial clefts have an estimated prevalence of 1 in 500 to 1 in 2500 newborns depending upon geographical, ethnic, and socioeconomic factors.1 Seventy percent of clefts are non-syndromic.1,2 Most non-syndromic clefts are presumed to be multifactorial in aetiology, with evidence indicating that genetic, environmental, and developmental determinants may all play a role.1,3 A few monogenic Mendelian disorders serve as models for non-syndromic clefting, such as Van der Woude syndrome at 1q424 and X linked cleft palate with ankyloglossia at Xq21.1-q21.3.5,6 EEC syndrome is another example of a Mendelian clefting syndrome. Recently, five families with EEC syndrome (OMIM 604292) were linked to chromosome 3q27, and heterozygous mutations were detected in the p53 homologue, p63, in nine unrelated EEC families.7 Another study has reported p63 mutations in two cases of EEC syndrome and two non-syndromic cases of ectrodactyly8 and recently the related disorder Hay-Wells syndrome has been shown to result from mutations in the SAM domain of p63.9
EEC syndrome is an autosomal dominant disorder with the cardinal signs of Ectrodactyly, Ectodermal dysplasia, and orofacial Clefts. Ectrodactyly, also called split hand/foot malformation, is a central reduction defect of the hands and feet that is often found with syndactyly.10,11 Ectodermal dysplasia can present as dry skin, sparse hair, dystrophic nails, or hypoplastic teeth, and lacrimal duct obstruction is often present as well.12 Clefting may affect the lip and/or palate.12 Other signs have also been associated with EEC syndrome. A review of 230 cases from English, German, French, Italian, and Dutch publications found EEC syndrome to include ectrodactyly (84%), ectodermal dysplasia (77%), cleft lip and/or palate (68%), lacrimal tract abnormalities (59%), urogenital abnormalities (23%), and conductive hearing loss (14%).13
The review of 230 cases identified 116 as familial and 114 as sporadic.13 Penetrance was estimated at 93% to 98% in this cohort.13 Highly variable expressivity has been reported for EEC syndrome.12–15 A comparison of interfamilial with intrafamilial variability in expressivity found a significantly larger interfamilial variability, suggesting that more than one gene or allele may be involved in EEC syndrome.13 In fact, two other loci have been proposed. Cytogenetic abnormalities implicate chromosome 7q11.2-q21.3.16–19 Linkage to chromosome 19 was found in a single Dutch family, Ams-1,20 but strong linkage for this same family was found for chromosome 3q27 as well.7 The linkage to one of these loci may be spurious, or, alternatively, a locus on chromosome 19 may act as a phenotypic modifier of p63.7
Nevertheless, causative mutations for EEC syndrome have only been identified in p63, a homologue of the tumour suppressor gene p53.7,8 Specifically, causative p63 mutations were detected in nine of 25 EEC families screened.7 Four additional mutations were reported, two in EEC patients and two in patients with isolated split hand/split foot8 and eight mutations were found in patients with Hay-Wells or AEC (ankyloblepharon-ectodermal dysplasia-clefting).9 A genotype-phenotype correlation was described for p63 mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand/split foot malformation.21 Expression of p63 is prominent in adult humans and mice in the proliferating epithelial basal cell layers of the epidermis.22 Embryonic expression in mice is prominent in the apical ectodermal ridge (AER) of the limbs and in the maxillary and mandibular branchial ectoderm, which are essential for the growth and patterning of the underlying mesenchyme.23,24 Homozygous p63 null mice exhibit craniofacial abnormalities, limb truncations, and absence of epidermal appendages such as hair follicles and tooth primordia, as well as absence of sebaceous, lacrimal, and salivary glands.23,24
In this report, we present heterozygous p63 mutations in three unrelated cases. Two cases are white families from Iowa. The other case is the sporadic case of a Filipino boy, the first report of a causative p63 mutation in a person of Asian descent. These cases exhibit significant interfamilial and intrafamilial variability in expressivity of the EEC syndrome.
MATERIALS AND METHODS
Subjects
Syndromic cases were identified at the University of Iowa Hospitals and Clinics and in the Philippines through Operation Smile. All cases had at least one affected family member examined by at least one clinical geneticist (JCM) or genetics clinical nurse specialist (SDH). Family histories were taken and blood or buccal swabs were collected for DNA analysis. Samples were obtained after signed informed consent following IRB approval both in the United States and in the Philippines. The identified cases consisted of five Iowa white cases and 34 Philippine cases. Previously described diagnostic criteria were used for EEC syndrome.13 Two of three cardinal signs must be present in isolated cases. In a familial case, one of three cardinal signs and a first degree relative with unequivocal EEC syndrome must be present to make the diagnosis. Also, related but separate syndromes were excluded from the diagnosis of EEC syndrome.13 Three of five Iowa white patients and one of 34 Philippine patients met these criteria for diagnosis of classical EEC syndrome. Cases of limb reduction defects and amniotic band sequence were also included in the study. A possible association of amniotic bands and EEC syndrome has been previously proposed.25 The clinical information for the cases is summarised in table 1. The data for cases in which mutations were detected are given in greater elaboration. Cases of non-syndromic orofacial clefts were also identified, 31 white patients through the Iowa Birth Defects Registry26,27 and 31 Filipino cases through Operation Smile.28
Clinical findings and syndrome designations of patients in the study
In case 1, three family members were affected in two generations of Iowa whites (fig 1). The affected family members (II.1, II.2, and III.1) all showed ectodermal dysplasia, with sparse to absent blonde hair, eyebrows, and eyelashes, dysplastic nails, palmar hyperkeratosis, and cone shaped teeth. All three also have renal abnormalities: an unspecified left renal abnormality (II.1), left renal agenesis (II.2), and right multicystic kidney and left dysplastic kidney (III.1). The proband (III.1) required a renal transplant following complications of peritoneal dialysis. The proband also has an isolated cleft of the soft palate, previously repaired. No other family member has a cleft and no family member has ectrodactyly nor lacrimal tract abnormalities. The father (II.2) does appear to have impairment of his lacrimal glands since he does not produce tears. The proband also has asthma requiring treatment with bronchodilators, chronic otitis media, and a repaired type I Arnold-Chiari malformation.
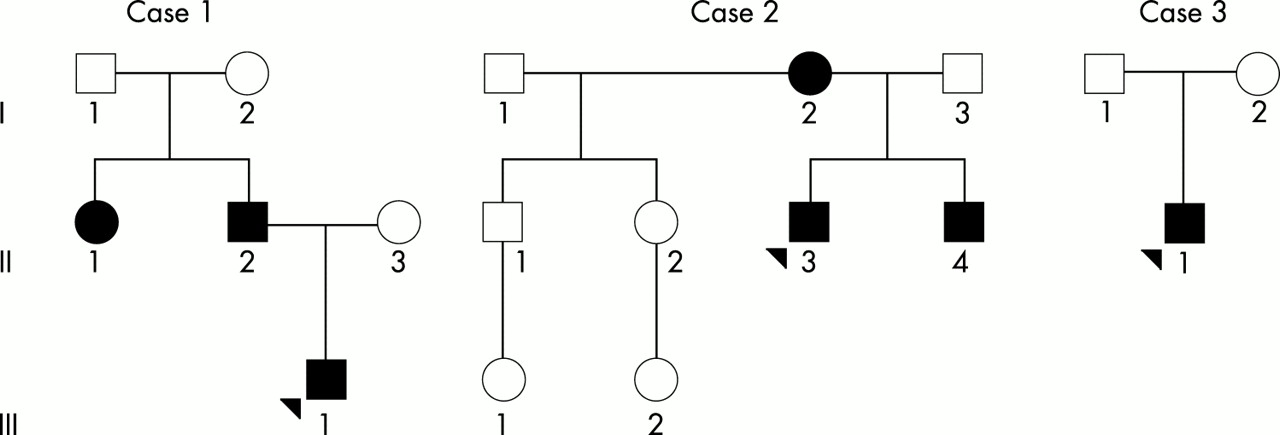
Pedigrees of EEC cases 1, 2, and 3. Affected subjects are indicated by blackened squares and circles. Probands are indicated by arrows.
In case 2, three family members were again affected in two generations of white Iowans (fig 1). The affected family members (I.2, II.3, and II.4) all show ectodermal dysplasia with sparse blonde hair, eyebrows, and eyelashes, dry skin, diminished perspiration, dystrophic nails, and peg shaped teeth with delayed dentition. The mother (I.2) is missing her central lower incisors, her first molar on the lower left side, and all wisdom teeth. All three (I.2, II.3, and II.4) have limb abnormalities, and both children (II.3 and II.4) have ectrodactyly. The mother has shortening of the first digits of her hands and fifth digits on her feet. The boys (II.3 and II.4) have missing digits 2-3 and 4-5 syndactyly of the hands bilaterally as well as missing right toes 2-4 and 4-5 syndactyly of the left foot. All three (I.2, II.3, and II.4) have lacrimal duct obstruction. The proband’s brother (II.4) has a bifid uvula. No other orofacial cleft was detected. The brother also had hypospadias. All three had clinically significant asthma requiring treatment with bronchodilators.
In case 3, the classical EEC triad occurred in the sporadic case of a Filipino boy (fig 1, fig 2). The boy had no sibs and neither parent was affected. He had bilateral ectrodactyly of the upper and lower extremities. He was missing digits 2-3 of the right hand and foot and missing digit 3 of the left hand and foot. He had sparse hair and hypodontia. He also had bilateral cleft lip and a cleft palate, which were subsequently repaired by Operation Smile. Lacrimal tract and urogenital abnormalities were not detected.
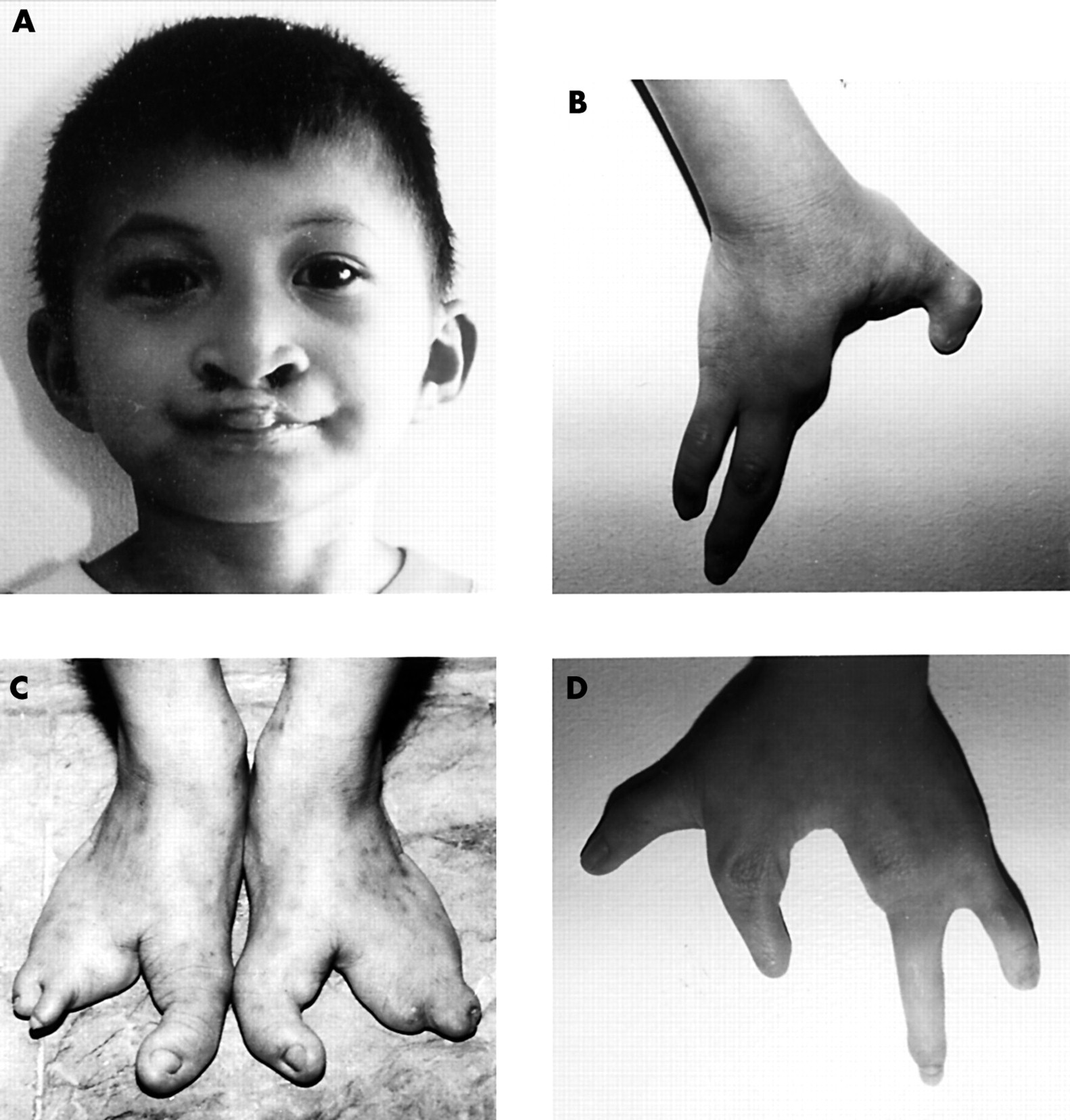
Clinical photographs of a sporadic case of EEC syndrome in a Filipino boy. (A) Bilateral cleft lip and sparse hair (shown); cleft palate and hypodontia (not shown). (B) Ectrodactyly of the right hand. (C) Bilateral ectrodactyly of the feet. (D) Ectrodactyly of the left hand.
Mutational analysis
SSCP analysis was conducted with 16 primer pairs external to each exon and inclusive of the splice donor and acceptor sites.7,29 Product sizes ranged from 140 to 366 bp, and products longer than 300 bp were digested with an appropriate restriction enzyme to produce two fragments less than 300 bp for SSCP analysis.29 PCR was conducted in a Stratagene Robocycler 96 in 10 μl reactions containing 1.5 mmol/l Mg2+, 40-80 ng DNA, Biolase Reaction Buffer (Bioline), 0.25 mmol/l each dNTP, 0.6 μmol/l each oligodeoxynucleotide primer, and 1.5 units Biolase DNA polymerase (Bioline). Cycling profile was one cycle of 94°C for three minutes, 40 cycles of 94°C for 40 seconds, 55-58°C for 30 seconds, and 68°C for two minutes, and one cycle of 68°C for seven minutes. Products were amplified in microtitre plates, digested when greater than 300 bp, electrophoresed on MDE gels, and silver stained.30 Variant and control bands were cut from the stained gel, amplified, and purified from agarose gel with QIAquick Gel Extraction Kit (QIAGEN). Genomic DNA was isolated as well. Purified products were sequenced bidirectionally with an Applied Biosystems (ABI) 373 DNA Sequencer using ABI Prism Dye. The 20 μl reactions contained 5% DMSO, 1.0 ng/μl DNA per 100 bp sequence, and 0.32 μmol/l oligodeoxynucleotide primer. Cycling was conducted in a GeneAmp PCR System 9700 (Perkin-Elmer). Cycling profile was one cycle of 95°C for five minutes, 99 cycles of 95°C for 20 seconds, 52-57°C for 20 seconds, and 62-67°C for four minutes. Sequence was analysed with Macintosh software including ABI Prism Sequencing Analysis 3.0 and Sequencher 3.1.1 (Gene Codes Corporation, Ann Arbor, MI).
RT-PCR
Total RNA was extracted from lymphoblastoid cell lines from affected subject III.1 of case 1 and from a control. cDNA was synthesised from 250 ng denatured RNA by random priming with random hexanucleotide primers (pd(N)6, Pharmacia) in a 20 μl reaction volume containing 5 mmol/l MgCl2, 10 mmol/l Tris base, 50 mmol/l KCl, 0.01% (w/v) gelatin, 1 mmol/l dNTPs, 2.5 U pd(N)6, 9 U RNA-guard RNase inhibitor (Pharmacia) and 5 U MMLV reverse transcriptase (Life Technologies). Reactions were 10 minutes at room temperature, 60 minutes at 37°C, and six minutes at 99°C. The integrity of the template RNA was checked upon electrophoresis in a 1.75% agarose gel by conducting a control RT-PCR experiment with IGF2R specific primers. For amplification of p63 alleles, we used exon specific primers matching to exon 8 (forward 8F) 5` - CAAGTTTCGGACAGTAC - 3`, exon 10 (forward; 10F) 5` - TGAAGATCAAAGAGTCCCTG - 3`, an exon 12 (reverse 12R) 5` - GACAATGCTGCAATCTGT - 3`, and exon 13 (reverse 13R) 5` - TCCATGGAGTAATGCTCAATCTG - 3`. The PCR reactions contained 5 μl of the synthesised cDNA, 62.5 ng of each oligonucleotide, 10 mmol/l Tris base, 50 mmol/l KCl, 0.01% (w/v) gelatin, and 1.25 U Taq polymerase (Life Technologies). Amplification was carried out in 35 cycles of 45 seconds at 95°C, 45 seconds at 50°C, and 90 seconds at 72°C. PCR products were analysed on 1.5% agarose gels. DNA fragments were excised from the gel and purified by use of the GEL Extraction kit (Qiagen). Both strands of the DNA fragments were sequenced with the above p63 specific oligonucleotides using the Big Dye terminator kit (PE). Sequence analysis was performed on an ABI3700 capillary sequencer (PE Applied Biosystems).
RESULTS
For p63 mutational analysis, we developed primer pairs external to each exon and inclusive of the splice sites based upon previous reports.7,29 SSCP was used to screen each proband for all 16 exons and all SSCP variants were directly sequenced. Additionally, all probands were directly sequenced for exons 5, 7, and 8 because these exons correspond to the highly conserved p53 DNA binding domain where 90% of the 10 000 mutations have been reported.7,31–33 Also, eight of nine previously reported p63 mutations were located in exons 5, 7, and 8.7
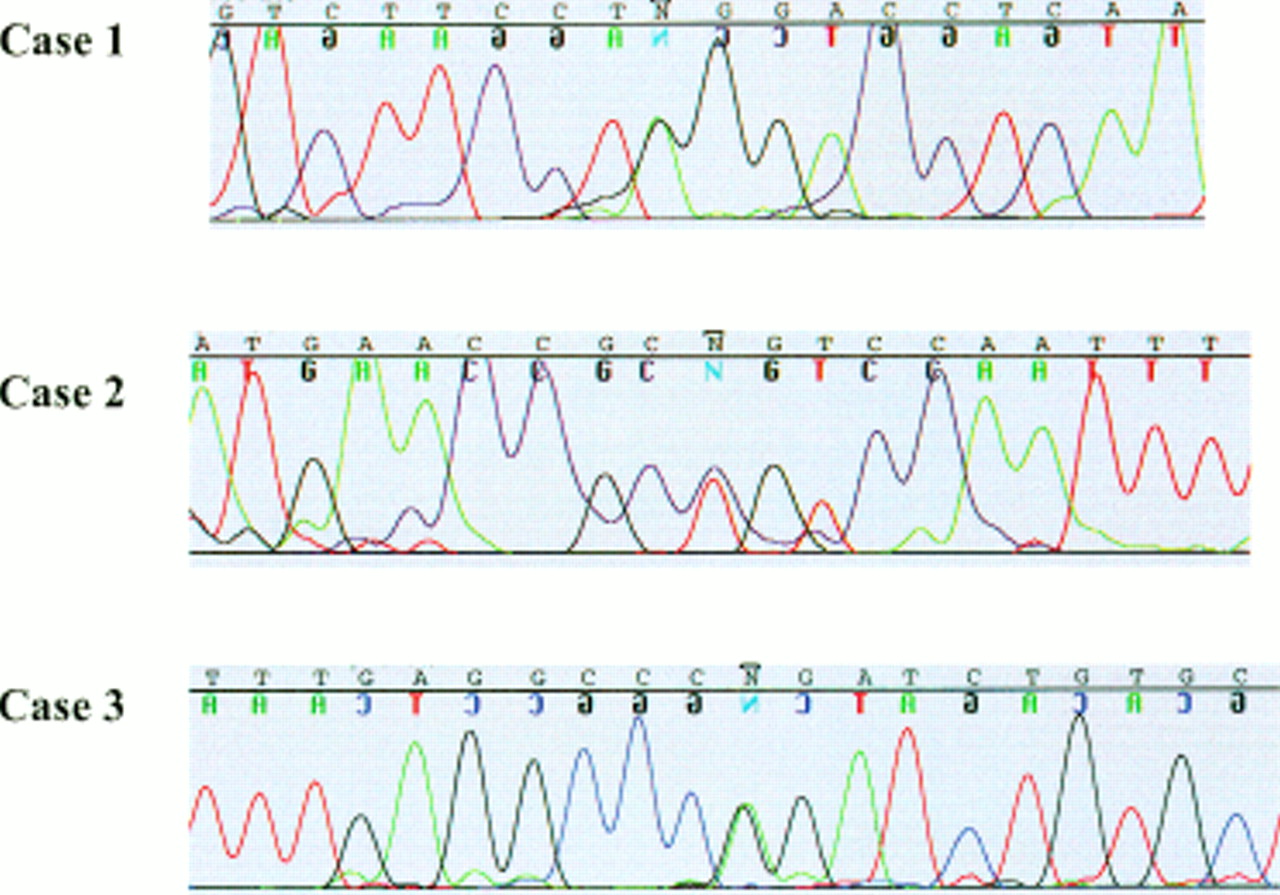
Heterozygous p63 mutations were detected in three unrelated cases of EEC syndrome, two by SSCP and one by direct sequencing alone (fig 3). The mutation in case 1, AG to GG, ablates the intron 10 splice acceptor site, directly preceding exon 11.34–36 Two missense mutations were detected, Arg280Cys and Arg304Gln (as numbered by the TA-p63α isotype). The CCG to CTG transition of Arg280Cys occurs in exon 7 of case 2 and the CGG to CAG transition of Arg304Gln occurs in exon 8 of case 3. Cosegregation of the mutant alleles with the affected family members of cases 1 and 2 was then confirmed. For case 1, family members II.1, II.2, and III.1 were heterozygous for the splice site mutation, and all other family members were homozygous wild type. For case 2, family members I.2, II.3, and II.4 were heterozygous for the Arg280Cys mutation, and the father I.3 was homozygous wild type. For case 3, proband II.1 was heterozygous for the Arg304Gln mutation, and his father I.1 was homozygous wild type. His unaffected mother I.2 was unavailable for testing. The three mutant alleles were not present in 200 control chromosomes. The position of mutations detected in EEC syndrome patients is illustrated in a schematic diagram of the human p63 gene in fig 4.

Chromatograms of heterozygous p63 mutations in cases of EEC syndrome. Case 1: AG to GG transition of splice acceptor site preceding exon 11. Case 2: CCG to CTG transition of exon 7 (R280C). Case 3: CGG to CAG transition of exon 8 (R304Q). Heterozygous nucleotides indicated by N.
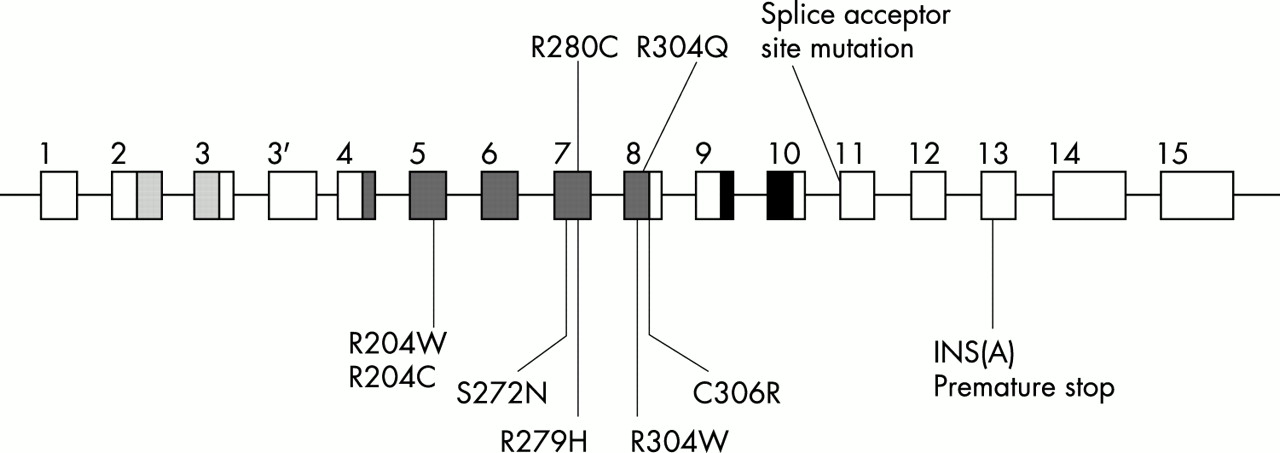
Schematic diagram of human p63 gene and mutations detected in cases of EEC syndrome. Intron-exon structure and conserved protein domains are shown.22 Transactivation domain: dotted. DNA binding domain: diagonal lines. Isomerisation domain: cross hatched. Previously reported mutations7 are indicated below and mutations reported in this study are indicated above. All missense mutations are located in the highly conserved DNA binding domain of p63 exons 5, 7, and 8. This conserved domain, analysed by crystallography for the p53 protein, forms DNA protein contact points and includes the most frequently mutated p53 residues in human tumours.7,31–33
All three mutations appear to be de novo. For case 1, the 274 bp allele of GATA6G12, a tetranucleotide repeat marker within 1 Mb of p63, was found to segregate with the splice site mutation, as seen in fig 5.7,37 The grandfather I.1 was found to carry the 274 bp allele but did not carry the splice site mutation in a buccal swab DNA sample, indicating that he may be a germline mosaic and the founder of this mutation.

Segregation analysis of GATA6G12 in case 1, an EEC family. GATA6G12 lies within 1 Mb of p63.7,37 PCR products were electrophoresed with CEPH controls at 60 W for three hours on a polyacrylamide gel. Size standards (in bp) are indicated on the right. The 274 bp allele segregates with the affected family members who carry the splice site mutation (II.1, II.2, and III.1). The grandfather I.1 carries the 274 bp allele but does not carry the splice site mutation in somatic tissue. Therefore, I.1 may be a germline mosaic and the founder of this de novo mutation.
Subjects with non-syndromic orofacial clefts were screened by SSCP and directly sequenced (table 2). A single nucleotide polymorphism was detected in intron 5 (+34 bp) in the white population. The genotypic frequency of the intron 5 polymorphism is T/T (20/31), T/G (7/31), and G/G (4/31). The intron 5 allelic frequency is then T (76%) and G (24%).
Mutational analysis of p63 exons in patients with non-syndromic orofacial clefts
The effect of the splice site mutation in case 1 was investigated by analysis of p63 specific transcripts from a lymphoblastoid cell line of III.1 and from a control. RT-PCR was performed with exon specific primers in three combinations: exon 8 to exon 13, exon 8 to exon 12, and exon 10 to exon 12, which for the control yielded products of the expected sizes (fig 6). The same products were also obtained for patient III.1, but in addition products of smaller sizes were observed. The sizes of these smaller products corresponded with the expected sizes of products resulting from skipping of exon 11. The integrity of the products was verified by sequencing. Indeed, the amplicons from the control as well as the corresponding amplicons from patient III.1 contained wild type p63 sequences. In contrast, the smaller product observed for patient III.1 contained p63 sequences without the entire 158 bp sequences of exon 11 (fig 6). Thus, the A>G splice site mutation is predicted to give rise to truncated p63 protein by the skipping of exon 11.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RT-PCR analysis of p63 transcripts in case 1 containing the 3` splice site mutation in intron 10. (Above) Analysis of RT-PCR products on a 1.75% agarose gel. p63 derived transcripts from lymphoblastoid cells of a control (normal) and patient III.1 (mutant). The amplified exons are indicated on top of each lane. The expected sizes of the wild type amplicons are on the left, while sizes of mutant amplicons are on the right. The aberrant RT-PCR products are indicated by an asterisk. A 100 bp ladder is used as a marker (M). A summary of the amplification reactions and product sizes is given in the diagram. Mutant alleles lack the 158 bp exon 11. (Below) Sequence analysis of excised bands from the gel. The top panel is the sequence of 406 bp product from exon 10 to exon 12 from the control, whereas the lower panel is the sequence of the 248 bp band obtained with the same primers with RNA template from patient III.1. The sequence clearly shows that this 248 bp product is the result of skipping of exon 11.
DISCUSSION
An AG to GG heterozygous mutation of the splice acceptor site preceding exon 11 was found in case 1. This family has features consistent with Hay-Wells syndrome and its mutation is unusual by location and type from the previously reported EEC and split hand/split foot cases.7,8 The previous reports of mutations in Hay-Wells syndrome all resulted from missense mutations in sterile alpha motif (SAM) encompassed by exons 13 and 14 and which might be affected by this frameshift in exon 11. Skipping of exon 11 results in a frameshift of the open reading frame. Transcripts lacking this exon could be easily detected in a lymphoblastoid cell line, indicating that mutant protein may be generated from it. Exon 11 is contained in only the α- and β-isotypes of p63, and deletion of this exon will produce truncated forms of these isotypes. A frameshift mutation resulting in a premature stop codon in exon 13 was identified previously in a patient with EEC syndrome.7 Transactivation studies indicated that this truncated protein exhibited dominant negative properties towards wild type p63 isotypes.7 A similar dominant negative effect is expected for the protein products generated by the exon 11 lacking transcripts, because the tetramerisation domain is still normally present. ΔN-p63α, the predominant p63 isotype in epithelial basal cell layers, lacks transactivation capacity for p53 reporter genes in vitro.22 However, ΔN-p63α does show prominent dominant negative activity to suppress the transactivation of these p53 reporter genes by both p53 and TA-p63γ.22 Transactivation studies with mutant p63 isotypes and p53 reporter genes in vitro suggest that hetero-oligomer formation between mutant and wild type p63 isotypes may explain the dominant negative activity of the p63 missense mutations in EEC syndrome.7 Since the RNA intermediate of the splice acceptor site mutation in case 1 is likely to be rapidly degraded in vivo, it begs the question as to whether this case might be an example of haploinsufficiency in EEC syndrome, as opposed to dominant negative activity. This question is at present under investigation.
The two heterozygous p63 missense mutations detected in this study, Arg280Cys and Arg304Gln, result in amino acid substitutions in residues which are highly conserved in sequence comparison of the DNA binding domains of p53, p63, and p73.7,22 The p63 DNA binding domain shares approximately 60% and 85% amino acid identity with p53 and p73, respectively.22 Arg280 and Arg304 of p63 correspond to Arg249 and Arg273 of p53, which are among the six most frequently mutated p53 residues.7,31–33 In the established nomenclature of p53 mutations, Arg304Gln is a class I mutation of the transcription factor p63 which disrupts protein-DNA contact points and Arg280Cys is a class II mutation of p63 which alters a residue essential for proper orientation of the DNA binding surface.32,33 A previous study has reported Arg304Gln in EEC syndrome and Arg280Cys in ectrodactyly.8 Transactivation studies of the mutated p53 proteins Arg249Ser, Arg273His, Arg273Pro, Arg273Cys, Arg273Leu, and Arg273Gly found all to reduce transactivation relative to wild type.38–40 Transactivation of the p63 missense mutant Cys306Arg is altered relative to wild type,7 but transactivation data for p63 missense mutants at Arg280 and Arg304 have not been reported.
In this study, we found heterozygous p63 mutations in three unrelated cases of EEC syndrome. Two cases are white families from Iowa, and the founder of one mutation appears to be germline mosaic. The other case is the sporadic case of a Filipino boy, the first report of a causative p63 mutation in a person of Asian descent. These cases exhibit significant interfamilial and intrafamilial variability in expressivity of the EEC syndrome, which is consistent with the reports of previous investigators.12–15 Variable ectodermal dysplasia was present in all three cases, but ectrodactyly, again with great variation, was found in only two cases. Familial EEC syndrome without ectrodactyly has been described previously.41 Alternatively, p63 missense mutations Lys194Glu and Arg280Cys have been found in cases of non-syndromic ectrodactyly, suggesting in a previous study that amino acid residues not directly involved in DNA binding might cause non-syndromic ectrodactyly as opposed to EEC syndrome.8 In this study, however, classical EEC syndrome with Arg280Cys was found in case 2. The affected family members of case 1 had renal abnormalities, which were not present in the other cases. Other clinical findings among these three cases included lacrimal tract obstruction, impaired lacrimal gland function, asthma, hypospadias, chronic otitis media, and Arnold-Chiari malformation.
Some form of orofacial cleft was present in a family member of each of the three cases. The proband in case 1 had an isolated cleft palate, but the two affected members of the previous generation did not have any cleft. Also, the brother of the proband in case 2 had a bifid uvula, but the proband and mother did not have a cleft. It is interesting to note that while the proband of case 1 had an isolated cleft of the soft palate and the brother in case 2 had a bifid uvula, the affected child in case 3 had bilateral cleft lip and a cleft palate. Genetics and embryology suggest that isolated clefts of the secondary (soft) palate may be a distinct clinical entity from clefts of the primary (hard) palate that involve the lip with or without the palate.42–44 The presence of both cleft types, coupled with the cardinal limb and ectodermal signs of EEC syndrome, implicate p63 as an early gene in development before an important bifurcation in palatogenesis. The extreme variation in expressivity of EEC syndrome, even within a given family, suggests that other genetic loci may play a significant role in the actual manifestation of the syndromic phenotype in any given person. Loci on chromosome 7q11.2-q21.3 and chromosome 19 are possible phenotypic modifiers. Alternatively, other as yet unidentified downstream factors may contribute. Certainly, the genetic factors contributing to orofacial clefts are heterogeneous, and p63 may elucidate downstream factors involved in non-syndromic orofacial clefts. In this study, we screened p63 in 62 patients with non-syndromic orofacial clefts, identifying an intronic single nucleotide polymorphism but finding no evidence of mutations that would explain even a subset of non-syndromic orofacial clefts. This study supports a common role for p63 in classical EEC syndrome, both familial and sporadic, but not in other related or non-syndromic forms of orofacial clefts.
Acknowledgments
We thank the families who participated in this study. We thank Operation Smile for support. We also thank Bonnie Ludwig, Sarah O’Brien, and Nancy Newkirk for technical support. This study was supported by grants DE08559 and DE11948 (JCM) from the US National Institute of Dental Research, and grant NWO 901-02-232 (HVB) from the Dutch Foundation for Scientific Research (NOW). Training was supported by US National Institutes of Deafness and Communication Disorders grant DC00040 (LLB), “Research Training Program for Otolaryngology”, administered by Dr Bruce Gantz.