Article Text

Abstract
Type I procollagen is a heterotrimer comprised of two proα1(I) chains and one proα2(I) chain. Chain recognition, association, and alignment of proα chains into correct registration are thought to occur through interactions between the C-terminal propeptide domains of the three chains. The C-propeptide of each chain contains a series of cysteine residues (eight in proα1(I) and seven in proα2(I)), the last four of which form intra-chain disulphide bonds. The remaining cysteine residues participate in inter-chain stabilisation. Because these residues are conserved, they are thought to be important for folding and assembly of procollagen. We identified a mutation (3897C→G) that substituted tryptophan for the cysteine at position 1299 in proα1(I) (C1299W, the first cysteine that participates in intra-chain bonds) and resulted in mild osteogenesis imperfecta. The patient was born with a fractured clavicle and four rib fractures. By 18 months of age he had had no other fractures and was on the 50th centile for length and weight. The proband's mother, maternal aunt, and grandfather had the same mutation and had few fractures, white sclerae, and discoloured teeth, but their heights were within the normal range. In the patient's cells the defective chains remained as monomers for over 80 minutes (about four times normal) and were overmodified. Some secreted procollagens were also overmodified but had normal thermal stability, consistent with delayed, but normal helix formation. This intra-chain bond may stabilise the C-propeptide and promote rapid chain association. Other regions of the C-propeptide thus play more prominent roles in chain registration and triple helix nucleation.
- osteogenesis imperfecta
- procollagen
- mutation
- carboxyl-terminal propeptide
Statistics from Altmetric.com
Assembly and triple helix formation of type I procollagen are stringently regulated processes. Type I procollagen, the soluble precursor of type I collagen, is composed of two proα1(I) chains and one proα2(I) chain, which are encoded by theCOL1A1 and COL1A2genes, respectively. Mutations in these genes disrupt normal procollagen biosynthesis and result in osteogenesis imperfect (OI), commonly known as brittle bone disease.1 Each proα chain contains a central obligatory Gly-Xaa-Yaa repeat sequence (helical domain) of more than 1000 residues (in which Xaa and Yaa are any residue other than cysteine or tryptophan), and globular propeptides at the amino- and carboxyl-termini. The helical domain is the functional part of the molecule that provides tensile strength and serves as a template for future mineral deposition in bone. The terminal propeptides are proteolytically removed from the molecule after secretion into the extracellular spaces, but play pivotal roles in the initial stages of procollagen folding and assembly in the cell. The C-propeptide directs chain-chain selectivity and assembly of the three proα chains before triple helix formation, yet little is known about the mechanisms that govern these processes.
The human proα1(I) and proα2(I) C-propeptides contain 246 and 247 residues respectively and are typical of globular proteins.2 3 They lack the obligate helical Gly-Xaa-Yaa repeat sequence and contain several cysteine and tryptophan residues, neither of which is found in the helix. The C-propeptides of proα1(I) and proα2(I) contain eight (C40, C46, C63, C72, C80, C151, C195, C242) and seven (C44, C67, C76, C84, C153, C198, C245) cysteine residues, respectively, the last four of which form intra-chain disulphide bonds.4 The other cysteines participate in inter-chain covalent bonds between chains in the same trimer. There is a complex pattern of inter-chain disulphide links between neighbouring proα1(I) and proα2(I) chains and intra-chain bonds between C80-C242 and C151-C195 in the proα1(I) chain, and C84-C245 and C153-C198 in the proα2(I) chain.4 These cysteine residues are conserved, consistent with crucial roles in the initial stages of procollagen assembly.5 6 The folded C-propeptide domain, stabilised by the formation of two intra-chain disulphide bonds, directs chain-chain selectivity, assembly of constituent chains, and proper chain registration, nucleation, and subsequent triple helix formation. However, it is not clear exactly what role, if any, the two intra-chain disulphide bonds play in these processes.
Proper chain selectivity ensures that each type I procollagen molecule has the correct composition of two proα1(I) chains and one proα2(I) chain. This is of particular importance when proα chains of different procollagens are coexpressed in the same cell. For example, human skin fibroblasts synthesise at least six precursor chains for fibrillar collagens, proα1(I), proα2(I), proα1(III), proα1(V), proα2(V), and proα3(V), necessary for the formation of procollagen types I, III, and V. Despite the high level of similarity, they assemble in a procollagen type specific manner. The ability to discriminate between chains is the result, at least in part, of a conserved discontinuous sequence of 15 residues in the approximate centre of the propeptide.7
After chain discrimination three C-propeptides assemble and promote correct registration of the triple helical domains. The helical domains of proα1(I) and proα2(I) are composed of 338 Gly-Xaa-Yaa triplets. Periodicity of charged and hydrophobic residues within this tripeptide along the collagenous domain provides opportunity for chain misalignment, but only one registration event results in correctly assembled molecules. The C-propeptides first associate through a series of non-covalent interactions that are then stabilised by the formation of inter-molecular disulphide bonds.8 Assembly of the C-propeptides in this fashion provides the correct alignment necessary for the initial nucleation event and helix propagation then proceeds in a C- to N-terminal direction along the length of the molecule.
We describe a naturally occurring mutation in the C-terminal propeptide of proα1(I) that results in the substitution of tryptophan for C80, thereby disrupting the intra-chain disulphide bond between C80 and C242. Rather than altering several residues, as seen in other frameshift mutations, this mutation replaces one cysteine with a tryptophan residue and leaves the remainder of the C-propeptide intact. This substitution slows the initial trimer formation, permits overmodification of residues within the triple helical domain, but in contrast to mutations in the triple helical domain, does not lead to destabilisation of the triple helix and has a mild phenotypic effect. These findings indicate that the cysteine is important for the rate of trimer association but that the phenotypic effects of the mutation are probably secondary to alterations in the post-translational modification of the triple helical domain, but not in helix stability. Further, these studies suggest that increased modification of the triple helix, per se, disturbs normal bone mineralisation.
Methods
CLINICAL HISTORY
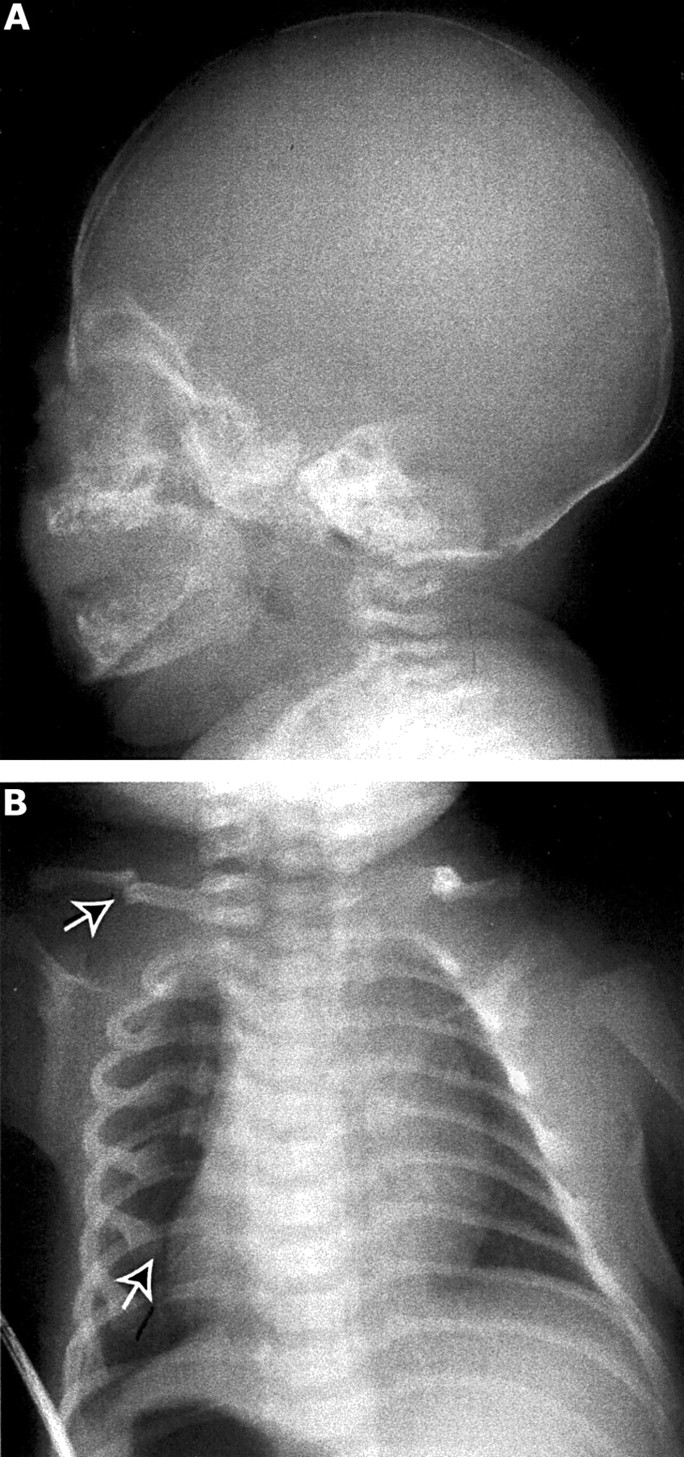
The proband (III.1, fig 1A) was identified at birth because of fractures of four ribs and a clavicle (fig 2B). He had wormian bones and an underossified cranial vault noted in a neonatal skullx ray (fig 2A). His birth length was 50 cm and weight 3402 g. At 2 years 7 months, he had had no additional fractures despite several opportunities, including falling down a flight of stairs and falling from his high chair. His length at 18 months was 80 cm (10th-25th centile) and at 2 years 7 months was 88.5 cm (5th-10th centile on the birth-3 curve, but between the 10th and 25th on the standard boys curve used for 1-18 year olds). His weight has continued to follow the 50th centile. Head circumference has followed the 25th-50th centile. His sclerae are white and he does not have macrocephaly or triangular facies. His teeth are slightly discoloured, but he does not have classical dentinogenesis imperfecta. He has somewhat soft skin.

OI producing mutation detected in three generations. (A) The index patient (III.1, arrow) was diagnosed at birth because of multiple fractures. (B) The mutation introduced a BstN1 restriction site, and restriction analysis showed that the proband's grandfather (I.1), mother (II.2), two aunts (II.3 and II.5), and cousin (III.2) also had the mutation. Clinical histories of two uncles (II.4 and II.6) and two cousins (III.3 and III.4), for whom biochemical and molecular studies were not completed, suggested that only one cousin (III.4) is affected (hatched).

X rays of index patient with OI. (A) In the perinatal period, there was diminished mineralisation of the calvarium and (B) fractures (arrow) of the ribs and clavicle.
His mother (II.2) is 1.55 m tall and has had one fracture (leg) sustained in a bicycle accident as a child. She had multiple caries and her teeth are yellowed. She has triangular facies, but no bone deformity. She has two sisters (II.3 and II.5) and one brother (II.6). One sister (II.3) is 1.65 m tall and has had several fractures, mostly fingers and a leg, related to trauma (volleyball, etc). She also had many caries as a child. Her husband (II.4) appears unaffected and they have three children. One daughter (III.2) of II.3 had three rib fractures and a clavicle fracture at birth and then had another clavicle fracture at about 6 months of age. She had wormian bones noted in a neonatal skull x ray. The other daughter (III.4) of II.3 had a single arm fracture at birth, but has yet to be tested. The other child has had no fractures. The second sister (II.5) has had no fractures but is 1.55 m tall. The brother (II.6) is 1.82 m tall and the only fractures were related to football trauma. The proband's maternal grandparents (I.1 and I.2) are alive and well. The maternal grandmother (I.2) is 1.60 m tall and the maternal grandfather (I.1) is 1.65 m tall. Both have had dentures since 19 years of age. Neither grandparent has hearing problems and neither has had significant fractures.
CELL CULTURE
Human dermal fibroblast cultures were established and maintained as described previously9 from biopsies obtained with appropriate consent. Before radiolabelling, cells were incubated with ascorbic acid (50 μg/ml) overnight.
METABOLIC LABELLING AND PULSE CHASE ANALYSIS
Fibroblasts were plated at a density of 250 000 cells per 35 mm dish and allowed to attach and spread overnight. Cells were preincubated for two hours in proline free medium and then labelled with [3H]proline (20 μCi/ml, Amersham Corp) overnight.9 For pulse chase experiments, the labelling medium contained 100 μCi/ml of [3H]proline and was aspirated after 80 minutes. Cells were rinsed with medium supplemented with 10% fetal calf serum and 10 mmol/l proline and then chased in the same medium. Labelled proteins from the medium and cell layers were harvested separately and ethanol precipitated in the presence of 15 mmol/l iodoacetamide to prevent new disulphide bond formation. Procollagens were partially digested with pepsin to generate collagens as previously described. For peptide mapping experiments, cells were labelled with 100 μCi/ml of [3H]proline overnight and then processed as described previously.9
DETECTION OF MUTANT SEQUENCE
Total RNA was extracted from cultured dermal fibroblasts using RNeasy Mini Kit™ (Qiagen). Complementary DNA was synthesised by priming with random hexamers in the presence of Superscript II™ RNase H- reverse transcriptase (Gibco BRL). Five large overlapping fragments of COL1A1 were amplified and analysed by SSCP as previously described.10A fragment including the region encoding the C-propeptide ofCOL1A1 was amplified by PCR using cDNA as the template. Primers 1A1X49S (sense) (5′-CAGATCGAGAACAT CCGGAGCCCA-3′) and 1A1X51A (antisense) (5′-CGGAGCCCTGGCCGCCATAC TCGA-3′) were used to amplify the region that encodes the C-propeptide domain ofCOL1A1. The PCR product was purified using QIA quick PCR Purification Kit (Qiagen). Sequencing reactions were then performed using the same primers and the ABI PRISM™ BigDye Terminator Cycle Sequencing Ready Reaction Kit according to the manufacturer's recommendations (Perkin-Elmer Applied Biosystems). Both strands were sequenced with the ABI PRISM™ 310 Genetic Analyzer. The mutation was confirmed in genomic DNA by directly sequencing the fragment using the same PCR primers listed above. Family members were screened for the mutation by isolating genomic DNA from peripheral blood samples using the PureGene kit as recommended by the manufacturer (Gentra Systems). PCR and sequencing of these subjects were performed using the same primer pairs. Presence of a mutation was confirmed by restriction analysis according to the manufacturer's recommendations.
HEAT DENATURATION
Tritiated procollagens were pepsin digested to form collagens, lyophilised, and resuspended in 50 mmol/l Tris/HCl, pH 7.4, containing 0.15 mol/l NaCl, 10 mmol/l EDTA, and 1% (v/v) Triton X-100. Samples were heated in a DNA thermal cycler from 36 to 43°C at a rate of 0.5°C every five minutes. At the end of each time period an aliquot was removed, cooled for one minute to 20°C, and digested with trypsin (100 μg/ml) (Sigma) and chymotrypsin (250 μg/ml) (Boehringer) for two minutes at 20°C, as described previously.11 The reaction was stopped by the addition of five volumes of SDS-PAGE sample buffer and boiling for three minutes. After electrophoresis, gels were processed for autoradiography and exposed to Kodak X-Omat AR film.
Results
OVERMODIFICATION AND POOR SECRETION OF TYPE I PROCOLLAGEN
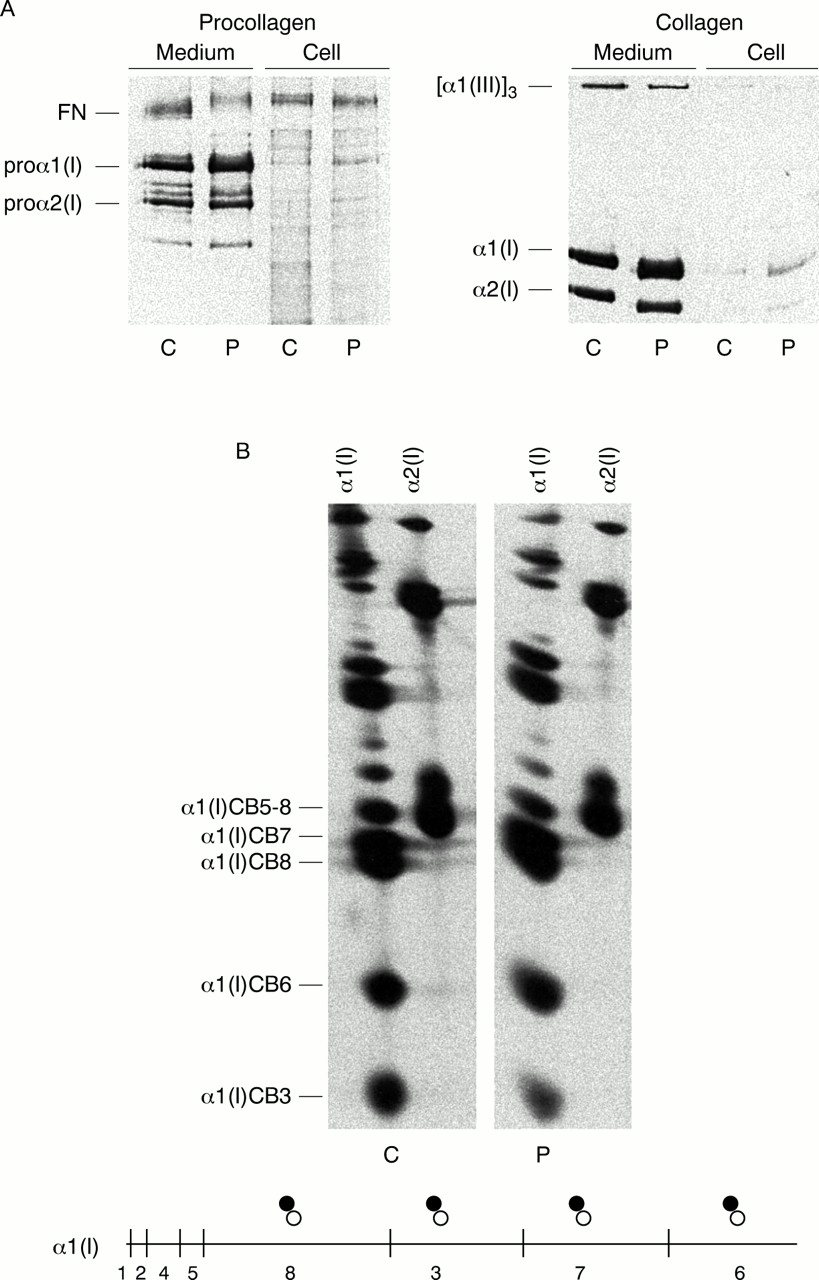
Cultured dermal fibroblasts from the patient synthesised and assembled some normal type I procollagen molecules along with a population of abnormal molecules that were pepsin resistant, but poorly secreted. The molecules that were retained intracellularly, and some of the secreted molecules, were composed of proα1(I) and proα2(I) chains that had retarded electrophoretic mobilities owing to excessive post-translational modifications (lysyl hydroxylation and hydroxylysyl glycosylation) (fig 3A). Two dimensional cyanogen bromide (CNBr) mapping of α chains indicated that overmodification extended along the full length of the triple helix, suggestive of a structural defect in the far C-terminal end of the triple helical domain or the C-propeptide (fig 3B).

Procollagen and collagen analysis. (A) Both control (C) and patient (P) cells synthesised normal populations of proα1(I) and proα2(I) chains, and OI cells also made proα1(I) chains with delayed mobility that were retained intracellularly. Collagens also had slowed electrophoretic mobility, suggesting that the helical domains were overmodified. (B) Peptide mapping of cyanogen bromide fragments showed that overmodification was distributed along the entire length of the helical domain. The diagram below the figure represents the α1(I) chain with the position of methionyl residues in the chain designated by vertical marks. The symbols above the chain designate the relative mobilities of the peptides derived from the normal chain (open circle) and the overmodified chain (filled circle).
MUTATION LOCATED IN THE C-PROPEPTIDE CODING REGION OFCOL1A1
Single strand conformation polymorphism analysis of cDNA detected a change in the C-propeptide coding region ofCOL1A1, consistent with a mutation in a single allele (data not shown). Direct sequencing of the fragment spanning exons 49 to 52 showed heterozygosity for a 3897C→G transition in exon 50 (fig 4A, B) that altered codon 1299 and predicted the substitution of cysteine by tryptophan (C1299W). The mutation introduced a BstNI restriction site. To determine the prevalence of this mutation in the proband's family, genomic DNA was isolated from several family members. Exon 50 was amplified by PCR and sequenced directly, and the mutation was confirmed by restriction analysis. The mutation was present in DNA from the maternal grandfather (I.1), the mother (II.2), two maternal aunts (II.3 and II.5), and a cousin (III.2) of the index patient (III.1) (fig1B).

Mutation characterisation. (A) Normal control and (B) patient cDNA was sequenced showing heterozygosity for a C to G transition of nucleotide 3897. (C) The mutation predicted the substitution of an evolutionarily conserved cysteine (1299) by tryptophan.
This cysteine residue is conserved in all the C-propeptides of fibrillar procollagens from human and other species (fig 4C). This cysteine forms an intra-chain disulphide bond with the last cysteine residue (C242 in the human sequence) in the chain and is thought to stabilise the domain before association with neighbouring chains.
DEFECTIVE C-PROPEPTIDE SLOWED CHAIN ASSOCIATION
Since the C-propeptide domain mediates chain-chain recognition and assembly of the proα chains into trimers, we examined the kinetics of procollagen assembly (fig 5). Fibroblasts were pulse labelled with [3H]proline for 80 minutes and chased for up to 80 minutes before protein isolation and separation by SDS-PAGE under non-reducing conditions. In control cells, most monomeric proα1(I) and proα2(I) chains were incorporated into dimers and trimers within 20-40 minutes, and the trimers were then secreted into the extracellular spaces. The patient cell strain synthesised two populations of proα1(I), a normal and an abnormal chain, along with normal proα2(I) chains. The normal proα1(I) chains showed the expected pattern of assembly and secretion by leaving the monomeric pool within 20-40 minutes and forming secretion competent trimers. The abnormal proα1(I) chains migrated more slowly through the gel, reflecting the extended formation owing to failure of C80-C242 disulphide bond formation, and were slow to assemble into dimers and trimers. They were retained within the cell for over 80 minutes and the electrophoretic mobility diminished through time, presumably because of further post-translational modifications. Also in these cells, secretion of type I procollagen trimers into the extracellular spaces proceeded more slowly than would be expected.
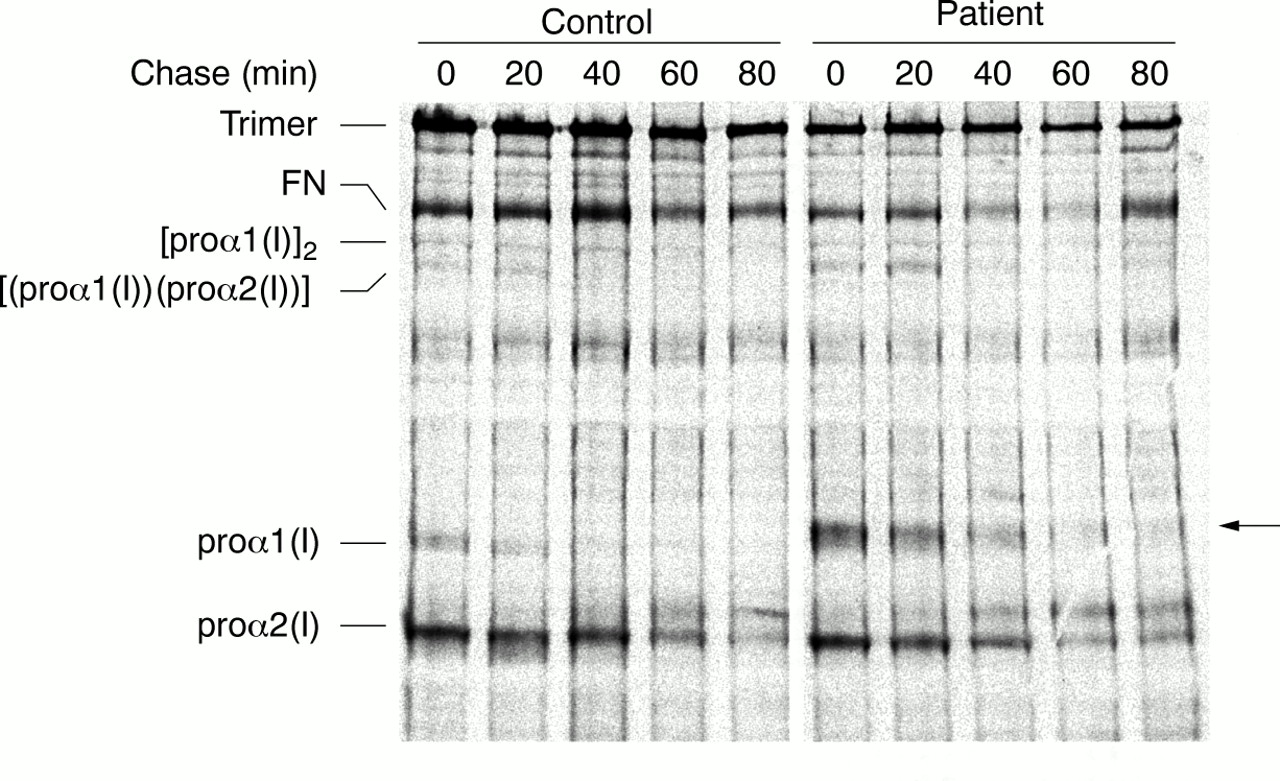
Pulse chase analysis of procollagen assembly and secretion. Whereas normal proα chains from both control and OI cells assembled into dimers and trimers within 20-40 minutes, defective proα1(I) (arrow) chains remained in the monomeric pool for over 80 minutes. Secretion of type I procollagen trimers was also slower than expected.
OVERMODIFIED TRIPLE HELICAL COLLAGENS WERE THERMALLY STABLE
Despite the excessive overmodification and the sluggish assembly and secretion of these abnormal procollagen molecules, thermal stability was normal (fig 6). The melting temperatures (Tm) of secreted collagen from both control and OI cells were equivalent and between 41.5 and 42.5°C. Thus, the C-propeptide defect and increased hydroxylation and glycosylation of the proα chains did not alter the structure of the triple helix in the way glycine substitutions do and, thus, did not decrease stability of the type I collagen trimer.

Thermal stability of abnormal molecules. Although markedly overmodified, abnormal procollagen molecules melted at the same temperature as normal molecules (41.5-42°C).
Discussion
We identified a point mutation in oneCOL1A1 allele that led to the substitution of tryptophan for cysteine at position 1299 (C80). Since the finding of the conservation of this residue, the disulphide bond that it forms is thought to play a crucial role in the initial stages of procollagen biosynthesis. Indeed, substitution of this cysteine by tryptophan and disruption of the disulphide bond that bridges C80 and C242 slowed procollagen assembly and secretion. However, in contrast to some other mutations that alter single residues in the C-propeptide and result in the lethal phenotype,12-15 the phenotypic effect of this substitution was mild OI with few fractures.
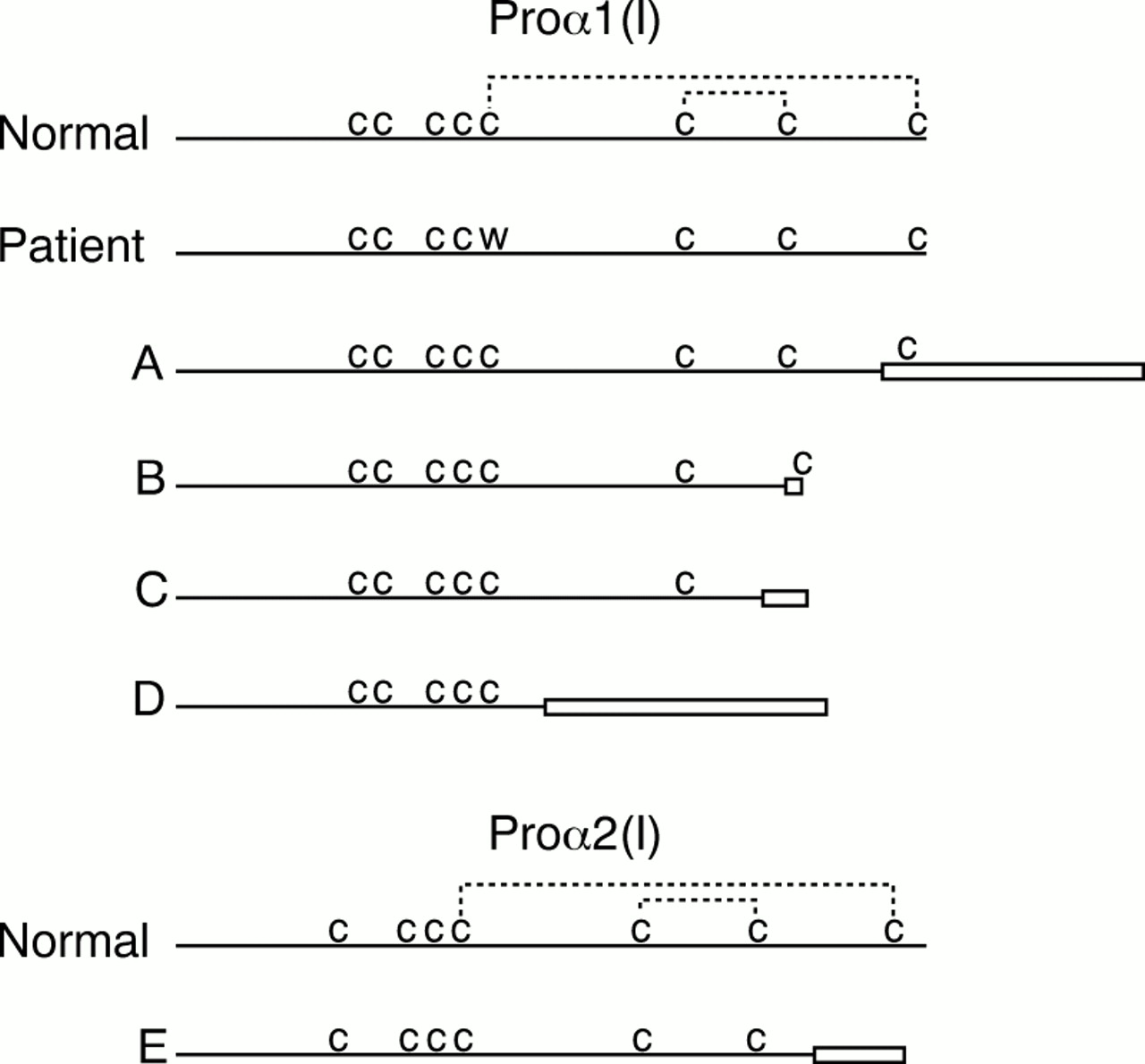
Of the more than 150 reported mutations in type I collagen genes that lead to OI, relatively few were found in the C-propeptide coding region, and fewer still disrupted disulphide bond formation (fig 7). A homozygous 4 bp deletion resulted in the synthesis of a proα2(I) chain in which the terminal 33 amino acids were altered, including the last cysteine. Similar to the presently described defect, the bond between C80 and C242 did not form. However, in that instance the alteration was more complex and abnormal chains were excluded from trimers, and the cells secreted proα1(I) homotrimers (fig7E5). A frameshift mutation inCOL1A1 that changed the translational reading frame and led to the synthesis of proα1(I) chains that extended 84 amino acids beyond normal termination (fig7A16) also changed the terminal cysteine residue, but these molecules were rapidly degraded, resulting in a functional null. Two other mutations in COL1A1 led to the synthesis of truncated proα1(I) chains without C195 and C242 (fig 7B, C). These two mutations disrupted both intramolecular disulphide bonds but, surprisingly, small amounts of pepsin resistant overmodified trimers were secreted into the extracellular spaces; the resulting phenotype was the perinatal lethal OI type II (J Pace, P Byers, unpublished data). Another mutation changed the reading frame and removed all four terminal cysteines (fig 7D14). The chains formed no intra-chain bonds but a small number were able to associate with other chains and form pepsin resistant molecules, most of which were retained in the cell. There was a marked decrease in the amount of type I procollagen secreted and the small number of abnormal molecules apparently led to a lethal phenotype. Although these naturally occurring mutations provide clues about the role of intra-chain disulphide bonds in the initial stages of procollagen assembly, it is difficult to separate the effects of abolished bond formation from the effects of other altered amino acid sequence in the C-propeptide.

Comparison of structural defects involving disulphide bonds of the C-propeptides of proα1(I) and proα2(I). There are eight cysteine (c) residues in proα1(I) and seven in proα2(I), the last four of which form intra-chain disulphide bonds (dashed lines). The others form bonds with neighbouring chains. Boxes represent new residues as a result of frameshift mutations. The mutation described here changed the first intra-chain cysteine to a tryptophan, preventing formation of the outside bond, and leading to slow assembly and little secretion. Other mutations resulted in more complex changes. Defects A16 and E5 precluded assembly into trimers. Defects B and C (J Pace, P Byers, unpublished data) resulted in slowed assembly with little secretion of defective molecules, and D14 permitted some assembly but no secretion into the extracellular spaces.
To understand better the effects of mutations that disrupt covalent bond formation in the C-propeptide, mouse proα2(I) chains in which one or more cysteine residues were substituted by alanine were expressed in a cell line that produced only endogenous proα1(I) chains. Single, double, or triple cysteine mutations were generated that disrupted one or both intra-chain disulphide bonds.17These molecules were assayed for their ability to associate and assemble with other chains and to be secreted as pepsin resistant molecules. Though secretion of detectable product was low, the data suggested that individual intra-chain disulphide bonds were not essential for assembly of the proα chains into trimers, only that at least one should form. This finding is at odds with previous studies,14 in which successful assembly of abnormal proα1(I) chains that lacked both intra-molecular disulphide bonds was described, which could mean that the structural requirements of the two chains differ.
Common among the mutations that alter intra-chain disulphide bonding is that they result in the slowed formation of overmodified procollagen molecules.18-20 The modifying enzymes, for example, lysyl hydroxylase, prolyl hydroxylase, and sugar transferases, remain associated with the unfolded proα chains and increase modification along the entire length of the chain but do not alter the placement of glycine residues or the length of the domain. In this way, the effects on the helical domain of mutations that alter the C-propeptide differ from those that alter the triple helical sequences themselves. In the latter, helix formation is thought to hesitate at the site of alteration, thereby allowing further modification of the chains from that point to the N-terminus. Although modification is increased, alteration in the relationships among the chains amino-terminal to the site of the mutation results in decreased stability of the trimer.
The studies we have completed are consistent with the following interpretation of the effects of mutation on the function of the altered proα1(I) chains. The substitution of tryptophan with a bulky side group for the compact cysteine residue may not interfere with the folding of the region distal to the alteration, but does prohibit the formation of the final stable product. In turn, this probably alters the shape of the chain recognition region that is located between the two cysteine residues and thus slows the access of altered chains into stable molecules until the mobility of the chain presents a reactive surface. Proper chain registration, helix nucleation, and triple helix formation are unaffected by the defect, but are delayed until chain association can occur. As a result of the delay, overmodification occurs because the chains remain associated with the modifying enzymes, an association that is terminated only by triple helix formation. It is likely that this intra-chain bond stabilises the C-propeptide in a conformation that promotes rapid chain selection and association. Other regions of the C-propeptide may mediate proper chain alignment and registration and promote helix initiation and formation.
Acknowledgments
This work was supported in part by a grant from the USPHS (AR 41223). James Pace is supported by the Molecular Training Program in Cancer Research (NIH T32 CA 09437).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}