Article Text

Abstract
BACKGROUND Rett syndrome is a neurodevelopmental disorder affecting only girls; 99.5% of Rett syndrome cases are sporadic, although several familial cases have been reported. Mutations in the MECP2 gene were identified in approximately 70-80% of sporadic Rett syndrome cases.
METHODS We have screened theMECP2 gene coding region for mutations in five familial cases of Rett syndrome and studied the patterns of X chromosome inactivation (XCI) in each girl.
RESULTS We found a mutation inMECP2 in only one family. In the four families without mutation in MECP2, we found that (1) all mothers exhibit a totally skewed pattern of XCI; (2) six out of eight affected girls also have a totally skewed pattern of XCI; and (3) it is the paternally inherited X chromosome which is active in the patients with a skewed pattern of XCI. Given that the skewing of XCI is inherited in our families, we genotyped the whole X chromosome using 32 polymorphic markers and we show that a locus potentially responsible for the skewed XCI in these families could be located on the short arm of the X chromosome.
CONCLUSION These data led us to propose a model for familial Rett syndrome transmission in which two traits are inherited, an X linked locus abnormally escaping X chromosome inactivation and the presence of a skewed XCI in carrier women.
- Rett syndrome
- skewed X chromosome inactivation
- X chromosome
- MECP2
Statistics from Altmetric.com
Rett syndrome (RTT, MIM 312750) is a severe neurological disorder affecting exclusively females.1 Its prevalence is about 1 in 15 000 live born females. Rett patients stop developing at about 1 year of age, and have a series of clinical signs indicative of a neurodevelopmental abnormality, including arrest of brain development, regression of acquired milestones, and behavioural troubles (stereotypic hand movements, autism).2 Most cases are sporadic (99.5%), although a few families have been reported. This pattern of inheritance is unusual and was until recently of totally unknown origin, thus making RTT one of the most exciting challenges in medical genetics. Several hypotheses have been proposed regarding the mode of inheritance and the mechanism by which Rett syndrome occurs. Male lethality has been proposed, but there is an apparently normal sex ratio in RTT families and no excess of miscarriages has been observed. Uniparental disomy has also been proposed, but after examination of Rett chromosomes, this hypothesis was ruled out. The most interesting suggestion, proposed by Thomas,3 is that the Rett mutation is male specific and can thus only be transmitted to females.
Another approach to elucidating the molecular mechanisms underlying this syndrome was to take advantage of the familial cases, in order to link the RTT locus to a particular region of a given chromosome. The X chromosome was first explored and several studies have excluded all of the X chromosome except the telomeric region of the long arm.4 5 Using cumulative data and multipoint analysis, it was shown that a RTT locus was linked to the Xq28 region, with a lod score of 2.9.6 For some families, it was shown that the mutation originated from the grandfather and was transmitted by unaffected carrier mothers.5
Mutations in the human methyl-CpG binding protein 2 (MECP2) gene were identified in 70-80% of sporadic Rett syndrome cases.7-9 We have thus screened the coding region of the MECP2 gene looking for mutations in five familial Rett syndrome cases and found aMECP2 mutation in only one, thus questioning the molecular basis of the Rett syndrome phenotype in the other four families.
One unusual observation in Rett syndrome familial cases is that carrier mothers transmit a pathological trait affecting only females and are not affected themselves, although the mutation possibly originates from their father's germline. One possible hypothesis for such reduced penetrance in carrier females would be the presence of a skewed pattern of X chromosome inactivation (XCI). Sirriani et al 6 reported a single RTT family in which an obligate carrier female had a completely biased XCI pattern, while her affected daughter exhibited a random XCI pattern. We thus focused our attention on the four families in which no mutation in theMECP2 coding region was identified. We assessed the XCI patterns in these families using the polymorphism of the CAG repeat of the androgen receptor (AR) gene (Xq12). Our results show that a totally skewed pattern of XCI segregates in the four families although it does not cosegregate with the disease. We genotyped the whole X chromosome in these families and we show that the only active X chromosome in the affected patients is the paternal X, although maternal transmission of RTT is likely to occur at least in one family. A potential controlling locus for this skewed XCI pattern could be located on the short arm of the X chromosome. These data led us to propose a model involving escape from normal inactivation of one (or several) X linked genes, and we ruled out MECP2 as being the gene potentially escaping X inactivation in the cases studied.
Methods
FAMILIES
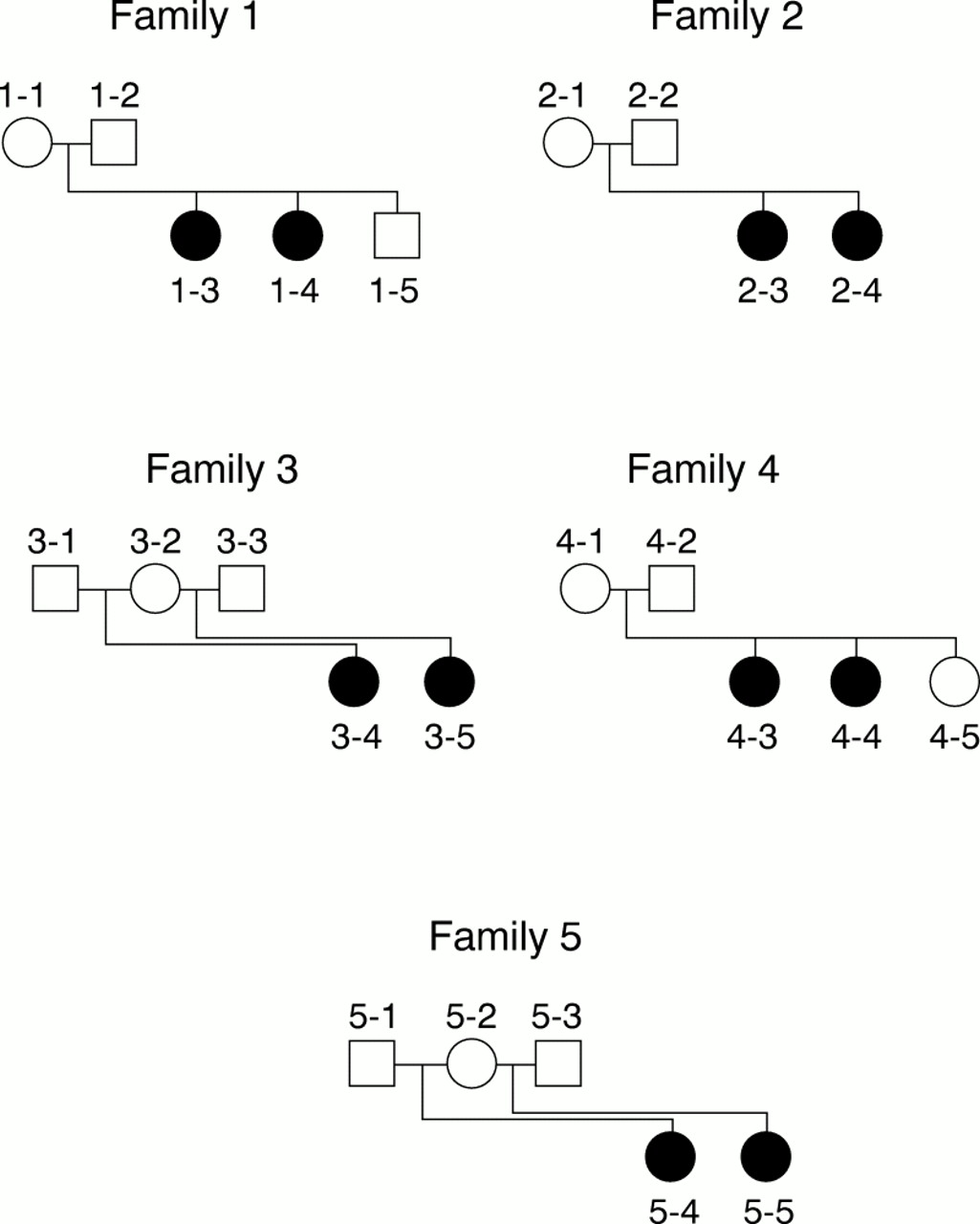
Families 1, 2, 3, and 5 in our report correspond to cases 5, 6, 2, and 3 of Xiang et al.5 Families 1 and 3 were reported by Hagberg et al,10 family 2 was reported by Haenggeliet al,11 family 4 has not been reported previously, and family 5 was reported by Ellisonet al.12 All affected girls fulfilled the international criteria for Rett syndrome diagnosis.
DNA SOURCES
DNA was extracted from lymphoblastoid cell lines according to standard procedures.
INACTIVATION STUDIES
Androgen receptor
Primers were designed in the (CAG)n flanking sequences of the androgen receptor (HUMARA) gene intron 1.13 Forward primer AR-P1 was 5′ labelled (Fam) and the reverse primer AR-P2 was unlabelled. Primer sequences were: AR-P1: Fam 5′ TCC AGA ATC TGT TCC AGA GCG TGC 3′; AR-P2: 5′ GCT GTG AAG GTT GCT GTT CCT CAT 3′. A total of 400 ng of DNA were digested by HpaII and ethanol precipitated. PCR reactions were performed with 100 ng of DNA both on HpaII digested and undigested DNA for all subjects. Males in our study were also tested under the same conditions for the allele size at the ARlocus after PCR on undigested DNA. For these males, PCR reactions onHpaII digested DNA were also used as digestion quality control (no amplification product onHpaII digested DNA at theAR locus). PCR conditions were as follows: 1 × PCR buffer, 0.2 mmol/l dNTPs, 1.25 mmol/l Mg++, and 0.5 UTaq (Gibco BRL) in a 20 μl final volume. Annealing temperature was 60°C for 30 cycles.
FMR1
Primers were designed in the (CGG)n flanking sequences of theFMR1 gene.14 Forward primerFMR1-P1 was 5′ labelled (Fam) and the reverse primer FMR1-P2 was unlabelled. Primer sequences were FMR1-F: Fam 5′-GCT CAG CTC CGT TTC GGT TTC ACT TCC GGT-3′, FMR1-R 5′-AGC CCC GCA CTT CCA CCA CCA GCT CCT CCA-3′.
PCR conditions were as follows: 1 × PCR buffer, 0.2 mmol/l dNTPs, 0.25 mmol/l 7-deaza-dGTP, 1.25 mmol/l Mg++, and 3 UTaq (Gibco BRL) in a 50 μl final volume. After initial denaturation for 10 minutes at 98°C, a hold step at 70°C was performed during which the Taqwas added to the samples, followed by one minute at 65°C and two minutes at 72°C. Thirty cycles were then performed for one minute each at temperatures of 98°C, 63°C, and 72°C. A final elongation step was done for 10 minutes at 72°C.
For both AR andFMR1 targeted assays, 1 μl of the PCR product was resuspended in 12 μl of TSR (Applied Biosystems). Samples were loaded on POP4 polymer on an automated ABI310 supplied with Genescan analysis software.
Controls
Inactivation studies were also performed with lymphocyte DNA from unrelated normal females as negative controls and XNP/ATR-X carrier women known to have a totally skewed pattern of XCI as positive controls.
SEQUENCING
The MECP2 gene was sequenced for each patient using genomic DNA as a template and also using cDNA for the patients for whom a lymphoblastoid cell line was available. Primer sequences for both genomic DNA and cDNA are available upon request. The PCR samples were directly sequenced, after purification with Qiagen PCR purification kit, using a LiCor sequencer and M13 tailed primers.
NORTHERN BLOTTING
Lymphocyte polyA+ RNA was prepared using Qiagen direct mRNA kit. One μg of RNA was loaded onto a 0.8% agarose 3% formaldehyde gel, and electrophoresis was conducted in 1 × borate buffer for six hours at 50 volts. The gel was transferred to HybondN+ membranes (Amersham) according to the manufacturer's instructions. Hybridisation was carried out in 50% formamide buffer at 42°C for 16 hours. A 800 bpMECP2 cDNA probe derived from the coding region of exon 3 was used to probe the northern blot.
RT-PCR EXPERIMENTS
Total RNA was extracted from lymphoblastoid cell lines using standard procedures.15 PolyA+ RNA was then prepared using the total RNA as a template using the Qiagen RNeasy kit (Qiagen) according to the manufacturer's instructions. The RNA sample was treated with RQ1 DNase for 15 minutes at 37°C and precipitated using standard procedures. One μg of polyA+ RNA was reverse transcribed in 50 μl of 1 × SuperScript reaction buffer (Gibco BRL) containing 3 ng/μl of dN6 oligonucleotides, 40 units of RNasin (Promega), 1 mmol/l dNTP, and 200 units of SuperScript II reverse transcriptase (Gibco BRL) for one hour at 42°C; 1/10th of this reaction mixture was subsequently used for PCR amplification.
GENOTYPING
The DXS markers used in this study are based on the Genethon human genetic linkage map. One primer for each PCR primer pair was labelled with the IRD800 infrared dye. PCR reactions were done in 50 μl volume for 30 cycles consisting of 45 seconds denaturation at 94°C, 45 seconds annealing at 55°C, and 45 seconds extension at 72°C. PCR products were directly loaded onto a LiCor automated sequencer to determine the allele sizes.
Results
FOUR FAMILIAL RTT CASES DID NOT HAVE MUTATIONS IN THE CODING REGION OF MECP2
We have studied five RTT families (fig 1) segregating a typical Rett phenotype fulfilling the international criteria for RTT diagnosis. We have screened these families for the presence of mutations in the coding region of the MECP2 gene using SSCP, direct sequencing of genomic DNA, and RT-PCR experiments using lymphocyte RNA as a template. RT-PCR products were normal in both size and abundance (data not shown), indicating that probably no mutation affecting either the splicing or stability of the mRNA was present in the families studied.

Pedigree of the five familial cases of Rett syndrome.
SSCP experiments and direct sequencing of genomic DNA did not show a mutation in the coding region of the MECP2gene in four families out of five. A missense mutation (C to T nucleotide change resulting in R106W) was found in one family (family 5) in the two half sisters but not in their mother. The presence of this mutation has been confirmed by restriction analysis of genomic DNA (data not shown) and was shown to be absent from 100 unrelated X chromosomes.
A TOTALLY SKEWED PATTERN OF XCI SEGREGATES IN THE RTT FAMILIES WITHOUT MECP2 MUTATION
In order to assess the XCI patterns in the four RTT families without MECP2 mutation, we used a polymorphism at the androgen receptor locus, and assayed methylation of the different alleles by digestion of the DNA with a methylation sensitive enzyme (HpaII) (fig 2). As positive controls, we used DNA samples from female carriers of the ATR-X syndrome since we have shown that a totally skewed pattern of XCI is caused by the presence of a mutation in the gene involved in this disorder.16

Transmission of the totally skewed XCI pattern in family 3. Numbering of subjects is the same as in fig 1. A totally skewed XCI pattern is observed both in the mother and her affected daughters after digestion with the methylation sensitive restriction endonuclease HpaII. The patient's active chromosome is of paternal origin in both cases while the maternally inherited chromosome is totally inactivated.
All the tested mothers (four out of four) had a totally skewed XCI pattern. In addition, six out of eight affected girls in the same families also have a totally skewed XCI pattern (the two affected girls not scored as totally skewed are 1-3 who displays a random XCI and 4-3 who is homozygous at the AR andFMR1 loci). In each case, we have scored the degree of skewing to >95:5 since no second allele was detected after amplification of HpaII digested genomic DNA on heterozygous samples (fig 2). Since lymphoblastoid cell lines were the only material available to perform this analysis, we postulated that our data could be the result of either the high proliferative status of lymphocytes or the transformation by EBV subsequently introducing an artefact into the results obtained. In order to rule out this possibility, which has been mentioned by other groups,8 we analysed the XCI pattern of 43 cases of sporadic RTT females. We decided to apply the same criteria as applied to familial studies and thus only skewed XCI with a ratio higher than 95:5 was scored as positive (that is, highly skewed). We found that 9% of RTT sporadic cases have a highly skewed XCI (table 1). In a next step, we analysed the XCI patterns of the mothers of the sporadic RTT females displaying a highly skewed XCI pattern. All the mothers displayed a random pattern of XCI, thus indicating that the skewed XCI is not transmitted from mothers to daughters and that it is thus probably not of genetic origin. In order to strengthen these results, we extended our analysis using lymphoblastoid cell lines from 52 CEPH families. We determined the XCI patterns of one female in each family. We found a skewed XCI pattern in 10% of females (as in the studied population of sporadic RTT cases). However, the skewed XCI pattern was transmitted from a mother to her daughters in one family among the 52 analysed. In our four RTT families withoutMECP2 mutation, the highly skewed pattern of XCI is segregating as a dominant genetic trait. A summary of the X chromosome inactivation analysis that we have performed is presented in table 1. In addition, we have tested the XCI pattern in the familial case in which we identified a mutation in theMECP2 gene (family 5) and we found that the mother had a random XCI pattern (data not shown).
Summary of the X chromosome inactivation tests performed on RTT mothers and their daughters (for the four familial cases without MECP2 mutation reported in this study), CEPH controls, sporadic cases of RTT, and ATR-X carrier females. Females were scored as “skewed” when the ratio of XCI was above 95:5
A CONTROLLING LOCUS FOR THE PRESENCE OF A SKEWED XCI PATTERN IS POTENTIALLY LOCATED ON THE PROXIMAL SHORT ARM OF THE X CHROMOSOME
To test if the skewed pattern of XCI observed in our four families is of genetic origin, we genotyped the whole X chromosome for each person using 32 polymorphic markers. For this analysis, we considered the skewed XCI pattern as being the “trait” and not the RTT phenotype. The results of this analysis are presented in fig 3. Exclusion mapping shows that almost all of the X chromosome can be excluded, excepted a region on the proximal short arm (Xp11.2-p11.4). This region has a genetic size of about 30 cM and a physical size of about 15 Mb based on the X chromosome genetic and physical maps, respectively. Multipoint linkage analysis was also performed using these data and a lod score of 1.5 was obtained with the same region of the X chromosome (data not shown). Additional similar familial RTT cases need to be added to the analysis in order to reach significance. We have haplotyped with the same markers the CEPH family in which we found that a skewed XCI pattern is transmitted from the mother to four of her five daughters, but the Xp11.2-Xp11.4 region was excluded by this analysis (data not shown).

X chromosome haplotypes obtained for informative subjects in the four families without MECP2 mutation. The healthy brother of the RTT patients in family 1 was not genotyped. Each parental chromosome is shaded with a different grey in order to show more clearly the recombinant chromosomes in the offspring. The pedigree representation was changed so that the trait (represented by black symbols) is the totally skewed XCI pattern. The critical region potentially containing the locus responsible for the skewing of XCI is located between DXS1068 and DXS1204 (horizontally shaded area).
IT IS ALWAYS THE PATERNALLY INHERITED X CHROMOSOME WHICH IS ACTIVE IN THE RTT GIRLS PRESENTING A TOTALLY SKEWED PATTERN OF XCI
The X chromosome haplotypes obtained in the previous step (see above) were used in order to determine the parental origin of the active X chromosome. In all informative girls with a totally skewed XCI pattern (six out of eight), it is the paternally inherited X chromosome which is active, the maternally inherited X chromosome being completely inactive (fig 4).

Parental origin of the active and inactive X chromosomes in the four Rett families studied for XCI. The androgen receptor (AR) alleles of each chromosome are indicated below each pedigree member. Circled AR alleles indicate a total bias in XCI resulting in 100% activity of the chromosome carrying this allele. The same representation is used for FMR1 alleles. Genotypes in Xq28 are indicated below FMR1 and the phase has been set between AR and Xq28 markers for each chromosome. Since the FMR1 locus was not informative in family 2, we were unable to determine its inactivation status.
Using the inactivation status at FMR1, the polymorphism of this locus, and the X chromosome haplotypes, we show that in families 1, 2, and 3 it is probably the same Xq27.3-q28 haplotype which is active in the mother and inactive in the affected girls who display a totally skewed XCI pattern. Concerning the haplotype associated with RTT, since the grandparents' DNA is not available for these families, and since all affected girls are obligate carriers of their father's haplotype, we cannot track which chromosome is carrying the Rett mutation. However, in family 3, it is very likely that it is a maternal chromosome that carries the RTT mutation since each patient has a different father.
ANALYSIS OF MECP2EXPRESSION
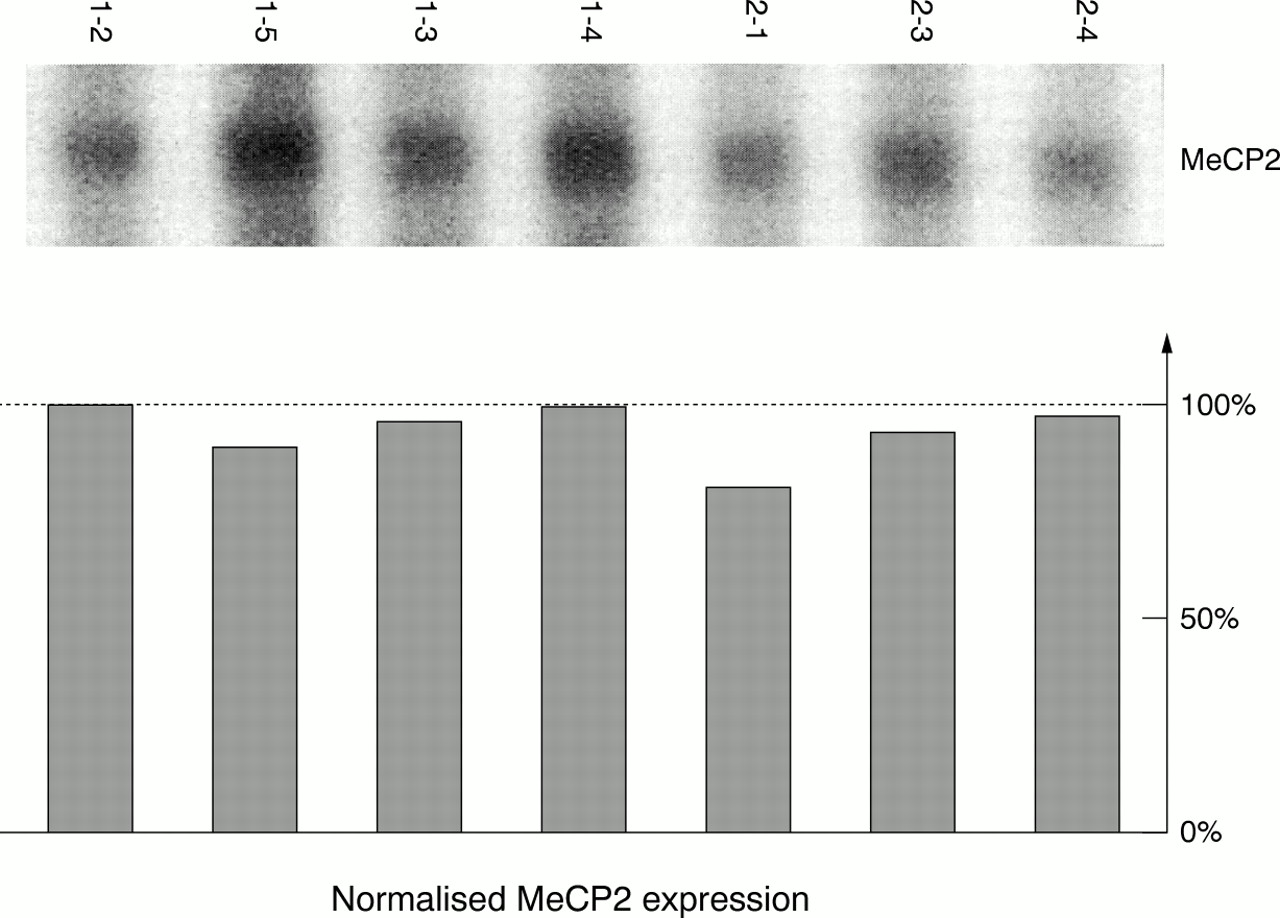
One explanation for the fact that 20-30% of the RTT sporadic cases do not have a mutation in the coding sequence ofMECP2 could be that other mutations affecting either the expression or the stability of the transcript are present in these subjects. In order to test this hypothesis, we performed northern blot analysis to quantitateMECP2 expression in the RTT families for whom cell lines were available. Normal males or females from the same families were also included in the analysis as controls. The results of this analysis are shown in fig 5. The amount ofMECP2 transcript in the affected girls is almost identical (maximal observed variation of 20% after quantification using phosphoimaging) to the amount present in control samples, thus probably ruling out the existence of a mutation affecting the expression of the gene or the stability of the transcript. However, a mutation which affected specifically the stability of the largeMECP2 transcripts (5 kb and 10 kb) would have been missed in our experiments since these two transcripts are poorly expressed in lymphocytes. The same experiments would need to be carried out using biological samples where the expression of these transcripts is high (for example, brain), which was impossible to perform in our families.

Analysis of MECP2 expression after northern blotting. PolyA+ RNA for each subject from two families (labelled as in fig 1) was hybridised with an MECP2 cDNA probe. (Top) MECP2 1.8 kb transcript expression. (Bottom) Quantification of MECP2 expression after analysis with a phosphoimager and correction of the MECP2 expression based on ribosomal RNA abundance (subject 1-2 was arbitrarily given a value of 100% for MECP2 expression); no variation larger than 20% was observed for MECP2 expression in these two families.
Another explanation for our results would be that the RTT phenotype in our four families without MECP2 mutation could be the result of a gene abnormally escaping inactivation (see Discussion) and this gene could be MECP2. Since the analysis of steady state mRNA levels using northern blotting can be tricky (especially if the dosage difference is 1v 2), we looked for a polymorphism in the 3′ untranslated region of MECP2, in order to determine the expression level of both alleles. We did indeed find a polymorphism in the MECP2 transcript after sequencing part of the 3′ UTR in the two affected patients in family 2 (G2356C, numbered according to GenBank entry AJ132917). Analysis of genomic DNA indicated that the “variant” allele was inherited from the father, the other being inherited from the mother (fig 6). The fact that the father is the carrier of the “variant” allele shows that the nucleotide change found in the 3′ UTR ofMECP2 is not disease causing. RT-PCR analysis using PCR primers flanking this polymorphism shows that the father expresses the “variant” MECP2allele (as expected) and that his two daughters are both expressing the same (paternal) MECP2 allele. No maternal allele was detected, consistent with the observation that both sisters have a completely skewed XCI pattern. This experiment shows thatMECP2 expression is monoallelic in this family, and that the RTT phenotype cannot be the result of a mutation causing MECP2 to escape inactivation.

Analysis of MECP2 expression in family 2 using RT-PCR. Results of the restriction analysis of a 358 bp PCR product containing the polymorphism identified in the 3′ UTR of MECP2. PCR products obtained from cDNA (left) or genomic DNA (gDNA, right) were digested with the BstNI restriction endonuclease. The wild type sequence contains four BstNI sites. The polymorphism identified in this family suppresses one BstNI site. The paternal allele is the only allele expressed by the affected RTT patients in this family. MWM=molecular weight marker. C+=undigested PCR product (there is a 40 bp difference between the undigested PCR product and the higher band after BstNI digestion).
Discussion
Our data show that a totally skewed XCI pattern segregates in the four RTT families that we have analysed. Strikingly, this skewed pattern does not affect the same X chromosome in mothers and in affected girls (since it is the paternally inherited X chromosome which is active in RTT girls). An analysis performed in normal subjects has shown that an extremely skewed XCI pattern can be found in females without any disease phenotype.17 In our hands, about 10% of the female lymphoblastoid cell lines (CEPH families or sporadic RTT cases) display a highly skewed XCI pattern. This can be because of either the high rate of proliferation of the lymphocytes or EBV transformation. However, this trait is not frequently transmitted through generations (one case out of 95 analysed in the present study), and is thus probably not of genetic origin. Hence, the segregation of a totally skewed XCI pattern in the four RTT families analysed in this study is probably not the result of chance, but rather an unknown biological factor. The fact that this trait segregates in our familial RTT cases strongly argues in favour of the transmission of a genetic trait. Two possible explanations for the presence of a skewed XCI pattern have been proposed. The first (“primary cause”) is probably linked to the mechanism of X chromosome inactivation in itself. This phenomenon has been observed in several families.18-20 In one case,18 it was linked to a mutantXIST allele. The second proposed mechanism is the selection against a deleterious allele during embryogenesis or cell differentiation. This is the case for several severe X linked disorders.21 22 In our study, the latter mechanism seems very unlikely for several reasons. First, it is always the chromosome carrying the mutation which is inactivated in carrier mothers of deleterious X linked traits and only males are affected, which is not the case in our RTT families. In addition, one would expect the Rett chromosome to be inactive in affected girls if a counterselection occurs against the deleterious allele. Thus, the first mechanism seems to be the most likely, as there is no need for the skewed pattern to be associated with a deleterious allele to be transmitted. However, when a skewed XCI pattern was identified, it seemed to affect the same X chromosome through generations and not to act randomly on maternal or paternal X chromosomes, as we observed in our families. At this stage of the work, it is difficult to explain the “inversion” of the skewed XCI pattern observed for the chromosome transmitted from mothers to daughters in our families.
If the totally skewed XCI pattern is a genetic trait segregating in these four RTT families, it should be possible to localise it by classical linkage analysis. Using 32 polymorphic markers distributed evenly along the human X chromosome for genotyping, we have excluded almost all regions of the X (including Xq28 and Xq13) from being the region in which a locus causing a completely skewed XCI pattern could be located. This shows that the two traits (that is, Rett syndrome and the completely skewed XCI pattern) are not linked, since the Xp11 region is excluded from involvement in the Rett syndrome phenotype based on allele sharing by affected girls in this region of the X chromosome. It also excludes a mutated allele ofXIST as being the cause of this completely skewed XCI pattern. However, a 30 cM region located between markers DXS1068 and DXS1204 systematically segregates with the skewed pattern of inactivation in our families and thus cannot be excluded. This region of approximately 15 Mb is a potential candidate region to contain a gene acting on X chromosome inactivation. Unfortunately, the lod score obtained using our data is not significant (Z=1.5 at θ=0) and more families need to be studied.
It had been proposed that the Rett locus could be inactivated in the mothers and active in their affected daughters.6 Our data rule out this hypothesis since it is the paternally inherited X chromosome that was found to be active in the affected daughters. In family 3, it is likely that the mother is carrying the RTT chromosome since the affected daughters have different fathers.
Our data lead us to propose a model in which the RTT mutation allows normally inactivated gene(s) to escape X inactivation. In the mothers, the chromosome carrying the RTT mutation is active, while the normal X is inactive leading to only one dose of the RTT gene(s) being expressed. In the affected daughters, the RTT chromosome is inactivated, but the mutant RTT gene(s) which escape(s) X inactivation remain(s) expressed. Since the paternal X chromosome is active, RTT affected girls will have two doses of the RTT gene(s) expressed (this mechanism is summarised in fig 7). According to this model, Rett syndrome would become familial when a mutation causing the RTT gene(s) to escape X inactivation occurs in a family in which a totally skewed pattern of X inactivation segregates, allowing the RTT mutation to be inherited. This hypothesis is compatible with the hypothesis of Thomas,3 stating that the RTT mutation originates from the father. A male specific germline mutation originating from the father or grandfather, which allows an X linked gene to escape inactivation, could thus be involved in sporadic (father's germline mutation) and familial (grandfather's germline mutation) forms of the disorder not resulting from MECP2 mutations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model for familial transmission of Rett syndrome. A hypothetical pedigree is presented with two parents and four children having received the “Rett” chromosome. The gene(s) escaping X inactivation and causing Rett syndrome is (are) represented by an open circle and arrows indicating its (their) expression irrespective of the XCI status. Totally inactivated chromosomes are shaded. Below the pedigree and for each child are indicated the percentage of activity of each chromosome, the status of the maternally inherited chromosome, the origin of the totally active X chromosome if applicable, the number of doses of the corresponding child, Rett gene(s) product(s), and the status of the corresponding child.
The MECP2 gene is involved in the control of gene expression through the binding of the corresponding protein to methylated CpG dinucleotides.23 These findings are of major interest since methylation is also a mechanism involved in X inactivation. According to our model, an alternative explanation is that the gene which escapes inactivation in our Rett families isMECP2. Unfortunately,MECP2 expression is the same in Rett girls as in their unaffected relatives based on northern blot quantification of MECP2 RNA. In addition, we tested the hypothesis of biallelic expression of MECP2in a family in whom we identified a polymorphism in the 3′ untranslated region of the gene. Only the paternal allele is expressed in his two affected daughters, ruling out that MECP2escapes inactivation in this family.
A possibility that should be mentioned is that we may have missed an unknown phenomenon which could affect MECP2, since lymphocytes were the only cells that we studied. A second hypothesis is that the molecular causes of Rett syndrome are diverse and thus it could be a genetically heterogeneous syndrome. In order to gain more information and to answer these questions, we are currently testing expression of the known genes in the linkage area, to detect a potential deregulation of one of them.
Acknowledgments
The first two authors contributed equally to this work. We would like to thank our colleagues and clinicians for providing the clinical material. We thank M J Mitchell, M G Mattéi, and F Muscatelli for helpful comments and discussions. We are grateful to Caroline Lacoste for excellent technical assistance. We thank Howard Cann for providing the CEPH families' DNA used in this study. This work was supported by the Association Française du Syndrome de Rett, Association Française contre les Myopathies, International Rett Syndrome Association, Axel and Margaret Axson Johnson Foundation, and the INSERM progress network.
References
1st Asia Pacific Forum on Quality Improvement in Health Care Three day conference Wednesday 19 to Friday 21 September 2001 Sydney, Australia
We are delighted to announce this forthcoming conference in Sydney. Authors are invited to submit papers (call for papers closes on Friday 6 April), and delegate enquiries are welcome.
The themes of the Forum are:
-
Improving patient safety
-
Leadership for improvement
-
Consumers driving change
-
Building capacity for change: measurement, education and human resources
-
The context: incentives and barriers for change
-
Improving health systems
-
The evidence and scientific basis for quality improvement.
Presented to you by the BMJ Publishing Group (London, UK) and Institute for Healthcare Improvement (Boston, USA), with the support of the the Commonwealth Department of Health and Aged Care (Australia), Safety and Quality Council (Australia), NSW Health (Australia), and Ministry of Health (New Zealand).
For more information contact:quality{at}bma.org.uk or fax +44 (0)20 7383 6869
Narrative Based Medicine, An Interdisciplinary Conference Research, Narrative, and Practice A two day conference—Monday 3rd and Tuesday 4th September 2001 Homerton College, Cambridge, UK
BMJ Publishing Group
For full details contact: BMA/BMJ Conference Unit, Tavistock Square, London, WC1H 9JP Tel: +44 (0)20 7383 6819; fax: +44 (0)20 7383 6663; email: clyders{at}bma.org.uk. www.quality.bmjpg.com