Article Text

Abstract
BACKGROUND We have previously described an autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia (MED), and distinctive facies in a large, extended Omani family. The MED observed seems to be part of a larger malformation syndrome, since both craniofacial and central nervous system changes were present in the family. We performed a whole genome scan in this family in order to identify the gene locus for this malformation syndrome.
METHODS AND RESULTS Using homozygosity mapping, we show linkage to the telomeric region of the long arm of chromosome 15. The position of both the disease gene and the principal glycoprotein, chondroitin sulphate proteoglycan (aggrecan, AGC1) on chromosome 15q26, suggested that the aggrecan gene is a candidate for the disease in this family. However, three of the four affected children were heterozygous for a polymorphism at position 831 of the coding sequence ofAGC1, providing strong evidence against its involvement.
CONCLUSION We have identified a gene locus for a recessive syndrome of macrocephaly, MED, and distinctive facies in a large Omani family. Aggrecan located on chromosome 15q26, within the critical region determined for this syndrome in this family, was excluded as a candidate gene.
- macrocephaly
- multiple epiphyseal dysplasia
- distinctive facies
- chromosome 15q26
Statistics from Altmetric.com
Multiple epiphyseal dysplasia (MED or EDM, OMIM 132400) was first described in 1947.1 It comprises a group of hereditary chondrodysplasias characterised by major anatomical abnormalities of the long tubular bones. The Fairbank and the Ribbing types are the most frequently cited types of MED and are defined radiographically.2 Both types are inherited in an autosomal dominant pattern. Common clinical findings are waddling gait, shortened extremities, genu valgum, and early onset degenerative joint disease. In one family with dominant inheritance, MED has been mapped to chromosome 19 and assigned as EDM13 (132400). It is caused by mutation in the cartilage oligomeric matrix protein gene4 (COMP, 600310). Since then many allelic variants of EDM1 have been described (600310). In a second family, also with dominant inheritance, the disease has been mapped to chromosome 15 (EDM2, 600304) and is the result of mutation in the COL9A2 gene (120260).5-7 In a third family, also dominant, the disease was mapped to chromosome 208 (EDM3, 600969) and results from mutation in the COL9A3 gene (120270).8 ,9
However, there have also been reports of MED families with a recessive mode of inheritance.10-15 In one family, there is evidence that autosomal recessive MED (EDM, 226900) can be caused by mutation in the diastrophic dysplasia sulphate transporter (DTDST) gene on 5q32-q33.1.16We report here an extended consanguineous Omani family with an autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia, and distinctive facial appearance, in which the disease gene has been mapped, using homozygosity mapping, to the telomeric region of the long arm of chromosome 15, adding another locus and further evidence for the genetic heterogeneity of MED.
Patients and methods
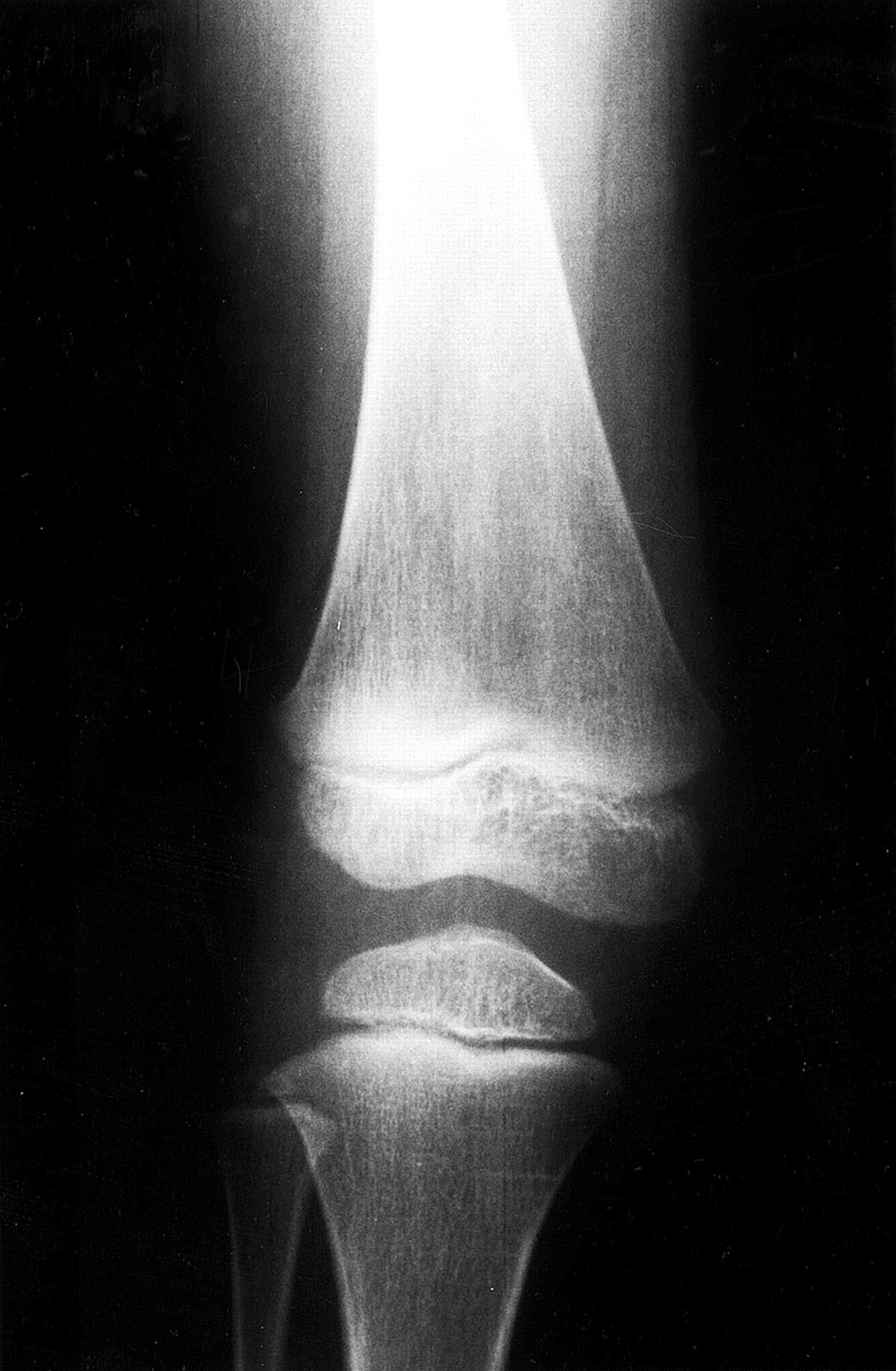
A large family of Omani origin has been described in detail elsewhere.17 The family shows multiple consanguinity. There are 13 children in two sibships and four of them are affected. The proband, a 10 year old male, and his two sibs, an 11 year old sister and a 5 year old brother, and their 2½ year old cousin were evaluated in the genetic clinic because of dysmorphic features, genu valgum, and swelling of some joints. All had normal height. Weight was above the 90th centile in three of them and head circumference was above the 90th centile in all of them. All four children had similar dysmorphic features consisting of macrocephaly with frontal bossing, hypertelorism, a flat malar region, low set ears (fig 1), a short neck, slight pectus excavatum, spindle shaped fingers with interdigital soft tissue webbing, and clinodactyly. All had genu valgum with prominent joints, particularly the wrist, knee, and ankle joints. Two had non-pitting oedema of the lower limbs. Skeletal survey on all four children showed dysplasia of the epiphyses of the long bones which was more severe in the lower limbs than the upper limbs (fig 2). CT scan of the brain on two of them showed agenesis of the corpus callosum and frontotemporal brain atrophy.

Facial appearance of an affected child. Note macrocephaly and frontal bossing. (Photograph reproduced with permission.)

X ray of the knee of patient 003 showing flattening of the femoral and tibial epiphyses. Note irregularities of the end plate of the femoral epiphysis.
GENOTYPING
Blood samples were drawn from all family members and DNA was extracted by standard methods. Microsatellite markers were chosen from the Généthon linkage map18 and labelled fluorescently. Amplification of markers was performed individually on MJ-Research Thermocyclers. The markers were pooled and electrophoresed on ABI 377 XL sequencers. Semi-automated genotyping was performed using the Genescan 2.0 and Genotyper V1.1 (Perkin-Elmer-ABI) software.
LINKAGE ANALYSIS
All genotypes were checked for Mendelian segregation with the help of the LINKRUN program (T F Wienker, unpublished data). All markers were scored for homozygosity in the patients. In addition, two point lod scores were calculated with the LINKAGE package19assuming recessive inheritance with full penetrance and equal allele frequencies for each marker. Multipoint lod scores (disease locus and three marker loci) were calculated with the program FASTLINK.20 ,21 Haplotyping was performed with Genehunter22 and manually. Marker data and genetic distances were taken from Dib et al.18
AGGRECAN-1 POLYMORPHISM
The human aggrecan proteoglycan (AGC1), located on 15q26.1, is a major component of cartilage. The chondroitin sulphate attachment sites in the human gene are made up of a variable number of tandem repeats (VNTR), which are highly polymorphic.23 We examined this polymorphism in all family members. The method used is essentially that of Doege et al.23 The primers used bracket the polymorphic site and amplify fragments 600-2300 bp in size. The 57 bp repeats were identified by size after agarose gel electrophoresis. In this family the number of VNTRs ranged between 27 and 29 repeats.
SINGLE NUCLEOTIDE POLYMORPHISM (SNP) ANALYSIS
The coding exons 2 to 18 of AGC1, with the exception of exons 4 and 12, were screened for SNPs using the Transgenomic WAVE DNA Fragment Analysis System (Transgenomic Inc, Santa Clara, CA, USA). This technique uses temperature modulated liquid chromatography and a high resolution matrix to distinguish hetero- from homoduplex DNA strands in heterozygotes.24 Briefly, 50 ng of genomic DNA were amplified using intronic primers flanking the respective exon. PCR products were heated to 95°C and cooled slowly to allow a mixture of homo- and heteroduplexes to form. Subsequent temperature modulated heteroduplex chromatography was used to distinguish homozygotes from heterozygotes. Primer sequences can be made available upon request.
SEQUENCING OF EXON 6 OF THE AGGRECAN (AGC1) GENE
Exon 6 was sequenced using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer Co, Foster City, CA, USA) on an Applied Biosystems 377 automatic sequencer.
Results
After scoring the markers for homozygosity by descent in the patients and performing parallel two point lod score analysis, we detected linkage to the long arm of chromosome 15. For the marker at locus D15S127, the two point lod score was Z=+3.45. Additional markers in the region confirmed evidence for linkage. Haplotype analysis showed that markers at loci D15S205 and D15S966 delimit the region in which all patients were homozygous while the unaffected sibs were heterozygous (fig 3). For the marker at locus D15S205, patient 003 shows a recombination giving a proximal border to the critical region. A distal recombination at the marker at locus D15S966 (subject 011, fig3) reduces the interval between the markers at locus D15S205 and locus D15S966. This leaves a homozygous region of 32.2 cM between the markers at locus D15S205 and locus D15S966. The remainder of the genome was excluded.

Pedigree of the Omani family with haplotypes on chromosome 15qtel for the markers D15S205, D15S127, D15S130, D15S1014, D15S120, D15S966, and the intragenic markers of the aggrecan (AGC1) gene. AGC1-6 and AGC1-12 represent exons 6 and 12 of the AGC1 gene respectively. Orientation of markers from centromere (top) to telomere (bottom) is given in the box on the top left hand side. Markers D15S1046 and D15S202 are included in the box to show the location of AGC1. Affected family members are denoted by black symbols. Black bars indicate disease linked haplotype.
EXCLUSION OF AGGRECAN-1 AS A CANDIDATE GENE
Several candidate genes have been suggested in the past for MED unlinked to known loci. Among these was the aggrecan-1 (AGC1) gene localised to 15q26.1.25 Aggrecan is a major structural proteoglycan of the extracellular matrix of cartilaginous tissue. A major function of aggrecan is to resist compression in cartilage. It contains a core protein of approximately 220 kDa,26 which is extensively modified by substitution with chondroitin sulphate, keratan sulphate chains, and other oligosaccharides.
We have tested the intragenic polymorphism of exon 12 of the aggrecan gene23 ,27 in this family. All four patients were homozygous for allele 27; however, not all unaffected sibs were heterozygous, since subject 005 was homozygous for the same allele. This rendered the AGC1 polymorphism uninformative in this family.
SNP analysis for the coding exons 2 to 18 ofAGC1 was also performed, with the exception of exons 4 and 12. The results exclude aggrecan as a candidate gene in this family as three of the four affected children are heterozygous for a polymorphism in exon 6 (C to G in position 831 of the coding sequence). The AGC1 gene has been mapped by two independent investigators using the GB4 Radiation Hybrid panel to the interval between D15S1046 (81.5 cM from ptel) and D15S202 (83.7 cM from ptel) (Genemap '99, http://www.ncbi.n1m.nih.gov/genemap/). As we have shown that the aggrecan gene is not shared identical by descent by three of four affected children (fig 3),AGC1 defines a new centromeric boundary to the critical region, leaving a homozygous region of 28.1 cM between the centromeric AGC1 marker at locus D15S1046 (81.5 cM from ptel) and the marker at locus D15S966 (109.6 cM from ptel).
Discussion
We have performed a whole genome scan with microsatellite markers spanning the genome with an average distance of 11 cM in order to localise the gene responsible for an autosomal recessive syndrome of macrocephaly, MED, and distinctive facies in a large extended Omani family. When typing markers of chromosome 15, we detected linkage to several markers on the q telomeric region. Two point lod score calculations for the marker at locus D15S127 resulted in a lod score of Z=+3.45, strongly suggesting genetic linkage of the disease gene to this region. D15S130, a marker distal to D15S127, also results in a two point lod score above +3 (table 1). Four point calculations gave a peak lod score of Z=+4.45 close to the marker at locus D15S1014 (fig 4). Haplotype analysis showed that all the affected children share the same haplotype, being homozygous by descent for the region between the marker at locus D15S205 and D15S966 (fig 3). These results give evidence for linkage of the disease gene in this family to chromosome 15q26.
Two point lod scores for markers on chromosome 15qtel

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Multipoint analysis performed with LINKMAP (from the FASTLINK package version 4.0P) with the markers D15S153, D15S205, D15S127, D15S130, D15S1014, D15S120, and D15S966, resulting in a maximal lod score of Z=+4.5 at marker D15S1014.
On considering candidate genes for the disease in this family, none of the 20 or more known distinctive collagen genes nor the cartilage oligomatrix protein genes were found within the critical region described in this family (Genemap '99). Other relevant genes situated close to the critical region are AGC1 andMANA2X, an isoenzyme of mannosidase alpha class 2A. The human aggrecan gene has been mapped to chromosome 15q26 by in situ hybridisation.28 ,29 Absence of this protein was observed in cartilage of mice that were homozygous for the autosomal recessive gene cmd (cartilage matrix deficiency).30 Watanabe et al 31 showed that a deletion in theAGC1 gene is causative for cmd in the mouse. Previous studies have ruled out the participation of the human aggrecan gene in autosomal skeletal dysplasias, such as achondroplasia or pseudoachondroplasia,27 but some types of achondrogenesis are phenotypically similar to cmd. We have tested the intragenic polymorphism of the aggrecan gene, a VNTR within the structural coding region of the gene,23 but it was found to be uninformative in this family. However, three of the four affected children were found to be heterozygous for a polymorphism in exon 6 (C→G in position 831 of the coding sequence). Heterozygosity of theAGC1 gene in affected children provided strong evidence against its involvement in this recessive syndrome.
The mannosidase alpha class 2A-X (MANA2X) isoenzyme is involved in the processing of N linked glycans, and has been cloned recently.32 It maps to human chromosome 15q26, between the markers at loci D15S202 and D15S127, just inside the critical region mapped for this recessive syndrome. The original isoenzyme, mannosidase alpha class 2 (MAN2A2), maps to chromosome 5q21-22. Its deficiency in humans is responsible for congenital dyserythropoietic anaemia type II. However, no deficiency disease has been assigned toMANA2X. Therefore, although an unlikely candidate gene, studies on MAN2X will be required to prove or disprove its involvement in this syndrome.
In summary, we have identified a gene locus for a recessive syndrome of macrocephaly, MED, and distinctive facies in a large Omani family. Aggrecan located on chromosome 15q26, within the critical region determined for the syndrome in this family, has been excluded as a candidate gene.
Acknowledgments
We thank Michaela Seeger and Mansour Qureshi for their excellent technical assistance, Franz Rueschendorf for statistical analysis, and Karl Sperling for his continued support. The financial support from the Deutscher Akeademischer Austausch Dienst (DAAD) to RB is gratefully acknowledged. The support by the Terry Fox Foundation, United Arab Emirates to MNEK is also acknowledged. The Gene Mapping Center is supported by a grant in aid from the German Genome Project to AR.
Electronic database information: accession numbers and URLs for data in this article are as follows: Généthon, http://www.généthon.fr(for information on microsatellite markers). Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for multiple epiphyseal dysplasia (MIM EDM1, 132400; EDM2, 600204; EDM3, 600969; EDM, 226900; AGC1, 155760; COMP, 600310; MAN2A2, 600988]). Genemap '99, http://www.ncbi.nlm.nih.gov/genemap/