Article Text

Abstract
BACKGROUND The GDNF family receptor alpha (GFRα) proteins are extracellular cell surface bound molecules that act as adapters in binding of the GDNF family of soluble neurotrophic factors to the RET receptor. These molecules are essential for development of many neural crest derived cell types and the kidney. Mutations in RET and in two members of the GDNF ligand family are associated with Hirschsprung disease (HSCR), a congenital absence of the enteric ganglia. Members of the GFRα family are also candidates for HSCR mutations. One such gene isGFRα-3, which is expressed in the peripheral nervous system and developing nerves.
OBJECTIVE We have characterised the structure of the human GFRα-3 locus and investigated the gene for sequence variants in a panel of HSCR patients.
METHODS Long range PCR or subcloning of PAC clones was used to investigateGFRα-3 intron-exon boundaries. A combination of single strand conformation polymorphism (SSCP) analysis and direct sequencing was used to investigateGFRα-3 sequence variants.
RESULTS GFRα-3spans eight coding exons and has a gene structure and organisation similar to that of GFRα-1. We identified three polymorphic variants in GFRα-3 in a normal control population, a subset of which also occurred in HSCR patients. We did not detect any sequence variants within the coding sequence of GFRα-3. We found a base substitution in the 5′ UTR of GFRα-3, 15 base pairs upstream of the translation start site. A second substitution was identified in intron 4 (IVS4-30G>A) between the splice branch site and the splice acceptor site. The final variant was a 2 base pair insertion within the splice donor consensus sequence of exon 7 (IVS7+4ins GG).
CONCLUSIONS We did not detect any correlation between variants of GFRα-3 and the HSCR phenotype. Our data suggest that mutations of this gene are not a cause of HSCR.
- GFRα-3
- Hirschsprung disease; RET
Statistics from Altmetric.com
The GDNF family receptor alpha (GFRα) proteins are a group of extracellular molecules, linked to the cell surface by a glycosyl-phosphatidylinositol (GPI) linkage.1-3 These proteins act as adapters which mediate the interaction of the receptor tyrosine kinase RET with members of the glial cell line derived neurotrophic factor (GDNF) family of soluble ligands. Four members of the GFRα family have been recognised to date, GFRα-1-4.4 5 GFRα family members share 30-45% identity and all have a similarly spaced arrangement of 28 cysteine residues, suggesting a conserved secondary structure.4 5Although structurally similar, each GFRα protein forms specific interactions with a subset of GDNF family members. For example, GFRα-1 is the preferred adapter molecule for GDNF,1-3GFRα-2 for neurturin,1 6 7 GFRα-3 for artemin,8 and GFRα-4 for persephin.9 GDNF family members cannot bind to RET directly. A GFRα family member first binds the GDNF family protein to form an intermediate complex which, in turn, binds to RET and triggers its activation. GFRα family members have distinct, although overlapping expression patterns. All are expressed, to some extent, in brain and peripheral and central nervous systems.4 5 However, the observation that mice lacking GFRα-1 or2 have significant but distinct phenotypes10 11 suggests that there are unique requirements for each of the GFRα family members in the normal developmental processes dependent on RET activation.
Disruption of RET signalling complexes has been implicated in the congenital abnormality Hirschsprung disease (HSCR).12 13In HSCR, the peripheral nerves of the gut fail to mature, resulting in absence of the myenteric nerve plexus along variable lengths of the gastrointestinal tract.14 The genetics of HSCR is complex; however, the RET signalling system contributes significantly to the phenotype. Germline inactivating mutations ofRET have been detected in approximately 40-50% of familial HSCR cases but in only 3-5% of sporadic cases in population based studies.15-17 Mutations of the RET ligands GDNF and NTN, which are hypothesised to abrogate RET signalling, have also been associated with the HSCR phenotype.18-21 To date, mutations ofGFRα family members have not been identified in HSCR,22 23 but not all members of theGFRα gene family have been investigated. Thus, these molecules remain candidates for mutations which could reduce or modify RET activity, resulting in HSCR.
GFRα-3 is highly expressed in the developing peripheral nervous system and decreases to low levels in the adult.24 25 GFRα-3 is expressed in the colon and small intestine and has been detected in neural crest derived cell types in the developing intestinal mucosa in early mouse embryos, suggesting a potential role in maturation.24 26 The early expression pattern in the gut and peripheral nerves and its demonstrated role in RET activation26 27 suggest thatGFRα-3 is a candidate for mutations in HSCR.
GFRα-3 has been mapped to 5q31.26 In this study, we have characterised the structure of the GFRα-3 gene and investigated the occurrence of mutations of this locus in a panel of patients with sporadic or familial HSCR. We show that the structure ofGFRα-3 is very similar to that of the previously characterised GFRα-1 gene. We identified three sequence variants at theGFRα-3 locus; however, our data suggest that mutations of GFRα-3 are not a common cause of HSCR.
Materials and methods
TEMPLATE DNA AND cDNA
DNA isolated from a panel of 36 unrelated HSCR patients, including 19 independent familial cases and 17 sporadic cases, was analysed in this study.22 DNA from 79 controls was also analysed. First strand cDNA was prepared as previously described28 29 from total RNA of the E293 cell line1 and used as template for RT-PCR.
PCR AND RT-PCR
PCR was carried out using a sense and antisense primer pair in 10 mmol/l Tris-HCl, pH 8.3, 50 mmol/l KCl, 0.75-1.75 mmol/l MgCl2, 0.01% gelatin, 1 μmol/l of each primer, 200 μmol/l each of dNTPs, and 1.5 U Taq DNA Polymerase (Life Technologies, Burlington, ON) with 40 cycles of one minute at 95°C, one minute at the primer specific annealing temperature (43-55°C), and one minute at 72°C, followed by a final extension at 72°C for 10 minutes. PCR products for sequencing were resolved on 2% LMP agarose gels and column purified using the Wizard PCR Preps kit (Promega, Madison WI).
PCRs for single strand conformation polymorphism (SSCP) analyses were prepared in 10 μl reactions as described above except that only 100 μmol/l dNTPs was used and 0.5 μCi of [α-32P]dCTP was added. Reactions were denatured at 95°C for 5-10 minutes and electrophoresed at 6 W overnight at 15°C on both 8% acrylamide and 6% acrylamide with 5% glycerol gels in 0.5 × TBE buffer. Two electrophoretic conditions were used in analysis of each PCR amplification in order to increase the efficiency of SSCP variant detection.
LONG RANGE PCR
Long range PCR was performed using the Elongase Enzyme Mix (Life Technologies, Burlington, ON) according to the manufacturer's instructions. Briefly, 200 ng of genomic or PAC DNA was used as a template in 50 μl reactions. Final concentrations of reagents included 1.6-1.9 mmol/l MgCl2, 200 μmol/l dNTPs, and 0.4 μmol/l of each primer. PCR amplification was performed for 40 cycles of 94°C for 30 seconds, 55°C for 30 seconds, and 68°C for three minutes. Products were resolved on 1.5% LMP agarose gels and purified (Wizard PCR Preps, Promega) as described above.
PAC CLONE CHARACTERISATION
PCR products corresponding to exons 5 to 8 ofGFRα-3 were used to screen a PAC library and two clones (273E22 and 282H7) were isolated (S Scherer, Hospital for Sick Children, Toronto, Canada). PAC clones were subcloned for further analysis to identify intron-exon boundaries. In brief, PAC DNAs digested with BamHI,TaqI, or PstI were ligated into pBluescript and the constructs electroporated into DH5α E coli using standard methods.30 Colonies containing inserts were gridded onto LB-ampicillin plates and colony lifts were performed using Hybond N+ membranes (Amersham Pharmacia Biotech, Baie d'Urfé, PQ) according to standard protocols.30 Membranes were hybridised to32P labelled exon specific PCR products or end labelled primers, as previously described.29 Positive colonies were isolated and DNA extracted using the Mini-Eclipse Plasmid Preps Kit (MoBio Laboratories, CA).
SEQUENCE ANALYSIS
Subclones and PCR fragments were sequenced using the Thermosequenase Kit (Amersham Pharmacia Biotech, Baie d'Urfé, PQ) either by incorporation of [α-32P]dCTP or using [α-33P]ddNTP terminators, according to the manufacturer's instructions. Reactions were resolved on 5% or 8% denaturing polyacrylamide gels.
PRIMERS
Primers used for SSCP analyses are shown. Coding sequence primers are shown in upper case.
Exon 1: L3F1 (5′-tctcagagctccaggggaggagc-3′), L3R1B (5′-ctcgtgcgccctctccggac-3′). Exon 2: L3F2D (5′-GAGACCCCCTTCCCACA GAA-3′), L3R2 (5′-CAAGGCTGCGGGC A CGGTGAACGGTC-3′); L3F2B (5′-GGAA CAGCTCTCTGATAGGCTGC-3′), L3R2D (5′-gccatcttccccagcttagtccactc-3′). Exon 3: L3F3B (5′-gcctaccccaccctggtcctca-3′), L3R3B (5′-cacccttcctccagggtccagcc-3′). Exon 4: L3F4C (5′-gaacctggggtgcaggcgt-3′), L3R4B (5′-CGGTCACACTTGTCATTGAGAG-3′); L3F4B (5′-GCGCCGGCGCAACACCATC GCC-3′), L3R4C (5′-agcacaggaacgtggccag-3′); L3F4D (5′-GCCATGCTGTGTACTCTCA ATG-3′), L3R4D (5′-AGTTGGGGGCGA TGGTGTTGCGCC-3′). Exon 5: L3F5B (5′-ggcaagagagagtgagaca-3′), L3R5B (5′-atctcc ggagcattcttac-3′). Exon 6: L3F6B (5′-ggtgga agaaggtgggagcta-3′), L3R6 (5′-TGAGG CAG GGGTTGTGGGAGAA-3′); L3F6 (5′-GGA CTGCCATGACCCCCAACTT-3′), L3R6B (5′-cagcctgggccacagagtcg-3′). Exon 7: L3F7B (5′-cttgggagcctgagaaatctcc-3′), L3R7B (5′-gcc tagtttgggttttcc-3′). Exon 8: L3F8 (5′-cccctg agctaactccattccatt-3′), L3R7 (5′-CTACCA TAGGCTCAGGAGCAGA-3′).
Additional primers used in identification of intron-exon boundaries and sequencing were selected using the reference cDNA sequence of Balohet al 31 (Accession number NM 001496).
Exon 1: L3F2 (5′-CGCCGTCGCCGCT GCCTCTCGC-3′). Exon 2: L3R2B (5′-GC AGCACTGCAGGTGGGATCA-3′). Exon 3: L3F3 (5′-GTAACTATGAGCTGGATGT-3′), L3R3 (5′-TGCTGAGATTCATTTTCCAG -3′). Exon 4: L3R4 (5′-TGCAAAGCGGGTC GGAGAAGCAGA-3′). Exon 5: L3F5 (5′-CAC GCCTGGTGGATTTCCAGA-3′), L3R5 (5′-CAATCAGCCCCAGGTATGCT-3′). Exon 7: L3F7 (5′-CGGAGGCCATTGCAGCTAAGA -3′).
Results
GFRα-3INTRON-EXON BOUNDARIES
The intron-exon boundaries of GFRα-3were identified using a combination of long range PCR and subcloning of PAC DNA fragments. The similarity of sequence and function among GFRα family members suggested that the gene structures might also be conserved. The intron-exon structure ofGFRα-1 has been previously reported.23 29 We estimated the approximate positions of intron-exon boundaries for GFRα-3 by comparison of its amino acid and nucleotide sequence with that ofGFRα-1. Forward and reverse primers were selected based on predicted boundaries. We compared the size of PCR amplification products obtained with genomic templates (PAC or genomic DNA) to those obtained from first strand cDNA generated from the E293 cell line. The presence of an intron was suggested by a larger PCR product with genomic templates, as compared to cDNA, with a given primer pair. Alternatively, GFRα-3 PACs were subcloned using several different restriction enzymes and exon containing clones were identified using labelled exon specific primers as probes. PCR products or clones were sequenced to identify the intron-exon boundaries and the contiguous intronic sequences (table 1).
Organisation of the GFRα-3 locus
We found that GFRα-3 spans eight coding exons varying in size from 89 bp to 313 bp (fig 1). TheGFRα-1 gene has 10 coding exons. Comparison of the structure of the two genes indicates that a small, alternatively spliced exon in GFRα-1 (exon 523 32) does not have a corresponding sequence inGFRα-3. Further, the final coding exon ofGFRα-1 (previously defined as exon 929 or exon 1123) is effectively absent inGFRα-3.

Schematic representation of the genomic structure of human GFRα-3 showing the location of sequence variants detected. Nucleotide changes for each site are given.
INVESTIGATION OFGFRα-3 IN HSCR
In order to investigate the occurrence of mutations ofGFRα-3 in patients with HSCR, we generated PCR primers flanking each exon for SSCP analysis. For larger exons, we analysed multiple overlapping PCR products in order to increase the efficiency of variant detection. To confirm putative sequence variants, we also investigated a panel of unrelated control subjects. We detected three variants in GFRα-3 (table 2, fig 2), each of which occurred in our control population. We did not detect any variants exclusively in HSCR patients. The nature of each variant was confirmed by direct sequencing. The positions of these variants are indicated relative to the GFRα-3 gene in fig 1.
GFRα-3 sequence variants in HSCR and control DNA samples

{kind=link}
{kind=link}
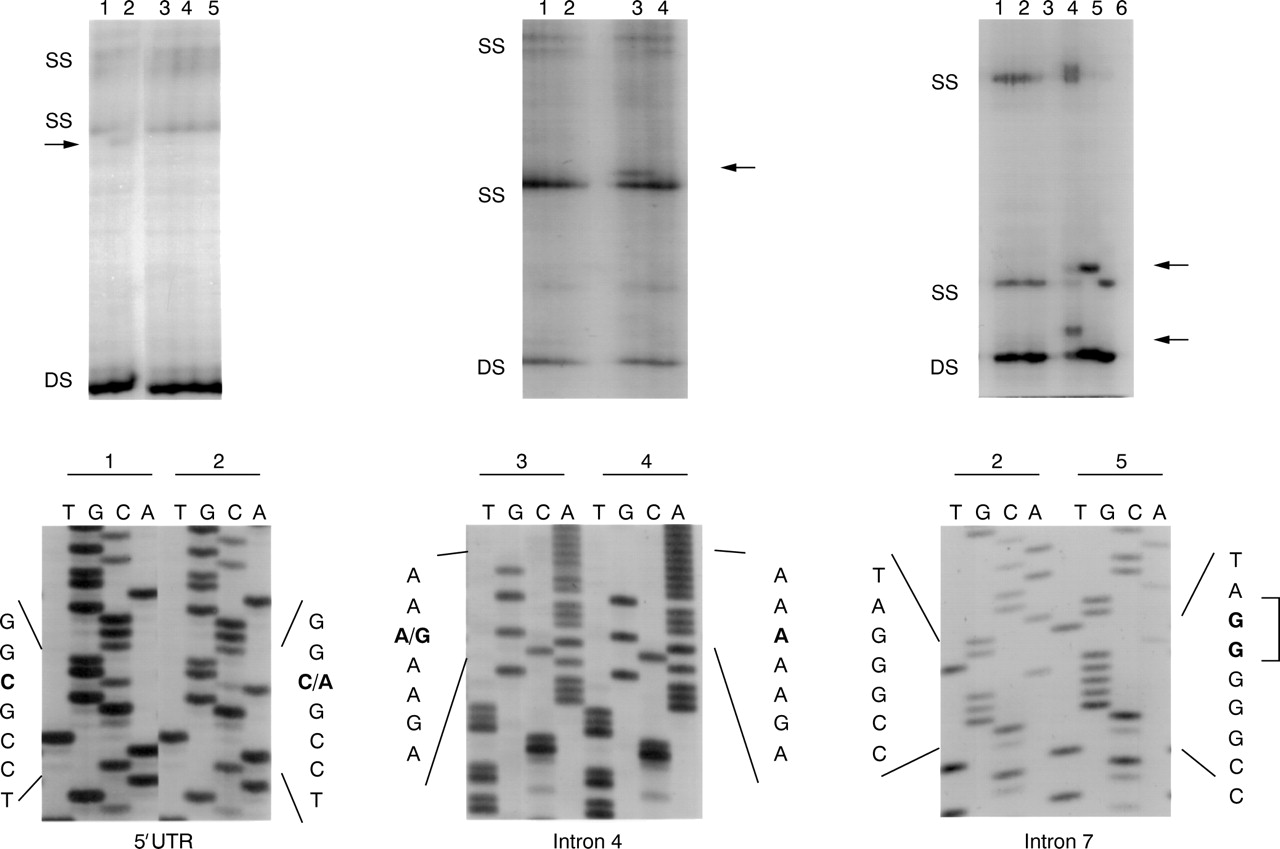
Sequence variants detected in GFRα-3. The SSCP patterns generated by variants in the 5′ UTR and introns 4 and 7 are shown. Single strand conformers (SS) corresponding to variant alleles are indicated. DS=double stranded DNA. Sequence showing the nature of each variant is shown below.
The three sequence variants detected all lie in non-coding sequences of the GFRα-3 locus. We identified an A>G substitution in intron 4 upstream of the exon 5 splice acceptor site (IVS4-30A>G) in both our control and HSCR populations. We identified two variants exclusively in our control population. The first of these was a C>A substitution within the GFRα-35′ UTR, 15 bp upstream of the translation start site (−15C>A). The second variant was a 2 bp insertion downstream of exon 7 (IVS7+4insGG). This sequence lies within the consensus splice donor recognition site (GTA/GAGT) changing it from a GTAGGGCC to GTAGGGGG.
Discussion
The GFRα family of adapter molecules is required to mediate the binding of the GDNF ligand family to the RET receptor tyrosine kinase. GFRα-3 preferentially binds artemin and the resultant complex can stimulate RET activation.8 TheGFRα-3 expression pattern is distinct from that of other GFRα family members and appears complementary to RET expression. In mouse, gfrα-3 is first detected at high levels at embryonic day E11 but expression declines subsequently to lower levels in adult tissues.25 27 33 Gfrα-3 is detected in the Schwann cells of developing nerves and in the trigeminal, dorsal root, and superior cervical ganglia, as well as some sympathetic ganglia.24 25 33 34 In non-neural tissues,gfrα-3 expression is highest in heart, lung, and in clusters of neuroendocrine cells in the developing intestine.24-26 35
Although structurally and functionally similar to the other members of the GFRα family, GFRα-3 is the most distantly related of the four family members. GFRα-1 and 2 share approximately 50% identity,1 6 7 36 while GFRα-3 has only 32 and 37% identity, respectively, with these proteins.26 35 Thus, it is not surprising that the gene structure is divergent from that ofGFRα-1. In this study, we show thatGFRα-3 has eight coding exons whileGFRα-1 has 10 coding exons.23 29 Several features ofGFRα-3 gene organisation were quite distinct from that of GFRα-1. InGFRα-1, a small 15 bp exon (exon 523 32) has been identified in a subset of alternatively spliced transcripts. Based on alignments ofGFRα-1 and 3, we would predict that a cognate exon would lie between exons 3 and 4 of the GFRα-3 gene (fig 1). We did not detect a corresponding exon in GFRα-3, nor could we detect a corresponding transcript using primers for the flanking exons in multiple tissue types (data not shown).
The sequences of GFRα-1 and 3 are most divergent near their C-termini.26 27 35 GFRα-3 is considerably smaller than GFRα-1 (400 versus 465 amino acids).26 27 33 Sequence corresponding to GFRα-1 exons 9, 10, and part of 11 are absent in GFRα-3 (fig 1). However, GFRα-3 is not merely truncated as the hydrophobic sequence involved in GPI anchoring25 26 is found in the final coding exon for each gene.
The expression pattern of GFRα-3 in peripheral nerves and in the neural crest derived cells of the gut24 suggest that it has a role in normal development and might be a candidate for mutations in HSCR. We investigatedGFRα-3 in a panel of 36 HSCR cases. All three of the sequence variants at theGFRα-3 locus were identified in our control population while only one of these was also found in our HSCR population. There was no significant difference in the frequency of variant alleles or genotypes between normal and HSCR populations for any of these variants. We detected a single base pair substitution (−15C>A) upstream of the translation start site which would not affect GFRα-3 protein sequence. It is possible that variants within this 5′ UTR region could affect the normal expression of GFRα-3 by disrupting promoter activity. However, this region did not contain any sequences known to contribute to transcription initiation. Analysis of these UTR sequences using the TF search database did not recognise any homologies to known transcription factor binding sequences which could have been disrupted by the C>A substitution. In intron 4, we identified a single base pair substitution 30 base pairs upstream of exon 5 and downstream of the splicing branch site. This change is not predicted to alter either the sequence or relative positions of the branch site and splice acceptor sequence and is thus not predicted to impact on the expression or function of GFRα-3. The final sequence variant detected at theGFRα-3 locus was an insertion of 2 bp in the splice donor site downstream of exon 7 (IVS7+4insGG). The consensus splice donor recognition site is GTA/GAGT. The two bp insertion would change the exon 7 splice donor site from AGgtagggcc to AGgtaggggg. This change is not predicted to alter the efficiency of splicing of this sequence.
Our data suggest that mutations ofGFRα-3 are not a common cause of HSCR. The absence of HSCR mutations in GFRα-3 is consistent with our previous observation that mutations ofGFRα-1 do not appear to contribute to HSCR.22 23 As mutations ofGDNF and NTN andRET are all associated with the HSCR phenotype, and as the GFRα family is required for RET activity, it is unclear why mutations of these family members do not contribute to the disease. Indeed GFRα-1 -/- mice, likeGDNF or RET -/- animals, lack enteric neurones throughout the intestine, a phenotype similar to very severe HSCR.10 37 It is possible that development of the enteric nerve plexus is not sensitive to the level of expression of the GFRα family while expression of RET or GDNF family members are limiting. Thus, reduced expression of RET, GDNF, or NTN could contribute to HSCR while mutations of a givenGFRα family member would not be associated with HSCR unless they completely eliminated the expression of that gene. Alternatively, it is possible that GFRα family members are more precisely regulated than are other components of the RET signalling complex in humans, and that mutation of anyGFRα family member which results in a reduction of functional product is associated with a more severe phenotype not recognised in our HSCR population. In recent studies,GFRα-3 null mice have been generated.34 These animals lack superior cervical ganglia, although neurones of other peripheral lineages appear grossly normal. A detailed study of the origin, function and density of the enteric neurones in these mice has not yet been reported. It is predicted that absence of GFRα-3 may affect migration and consequent survival of some neural crest lineages.34 We did not identifyGFRα-3 mutations in patients with HSCR alone or in patients having HSCR with classical associated phenotypes such as multiple endocrine neoplasia type 2, Down syndrome, or pigmentary abnormalities. Together, these data indicate that mutations of GFRα-3 do not lead to HSCR. However, current interpretation of genetic data suggests that HSCR is multifactorial in nature, with variants such as polymorphisms at several loci contributing to expression of the disease phenotype.18 20 21 38-40 Our data suggest that sequence variants of the GFRα-3 coding sequence are not likely to represent common modifiers of the HSCR phenotype. Future studies will need to examine the effect of variants outside the GFRα-3 coding sequence, or at other GFRα loci, on this phenotype.
Acknowledgments
We gratefully acknowledge the patients and families, their clinicians and geneticists who contributed so greatly to this study. We thank Dr S Scherer for PAC clone isolation and C Hession and D Worley (Biogen Inc) for providing sequence information. We are grateful to Drs R Hofstra and G Cote for helpful discussions. This work was supported by grants from the Medical Research Council of Canada and the Hospital for Sick Children Foundation.