Article Text

Abstract
METHODS A large family is described in which mental retardation segregates as an X linked trait. Six affected males in three generations were studied by linkage and clinical examination.
RESULTS Characteristic clinical features include short stature, prominent lower lip, small testes, muscle wasting of the lower legs, kyphosis, joint hyperextensibility, abnormal gait, tremor, and decreased fine motor coordination. Affected subjects also had impaired speech and decreased attention span. A carrier female was mildly affected. A similar disorder was not found on review of our XLMR Database of 124 syndromes. Linkage analysis of 37 markers resulted in a lod score of 2.80 at DXS1212 and 2.76 at DXS425. The limiting markers were DXS424 and DXS1047. Ten of 124 XLMR syndromes and eight of 58 MRX families overlap this region.
CONCLUSIONS In summary, this family appears to have a new XLMR syndrome localising to Xq24-q25.
- X linked mental retardation
- Xq24-q25
- syndrome
Statistics from Altmetric.com
In the two year interval between the “XLMR Gene Updates” of 19961 and 1998,2 the total number of known syndromes increased from 105 to 120 and the reported families with non-specific XLMR (MRX) increased from 42 to 60. During this time the cloning or molecular characterisation of these disorders has resulted in the “lumping” of disorders found to be allelic, so that the overall increase in the number of syndromes represents the net result of both new reports and the “lumping” of allelic disorders. We report here the clinical findings and linkage studies of another new syndromic form of XLMR.
Subjects and methods
FAMILY REPORT
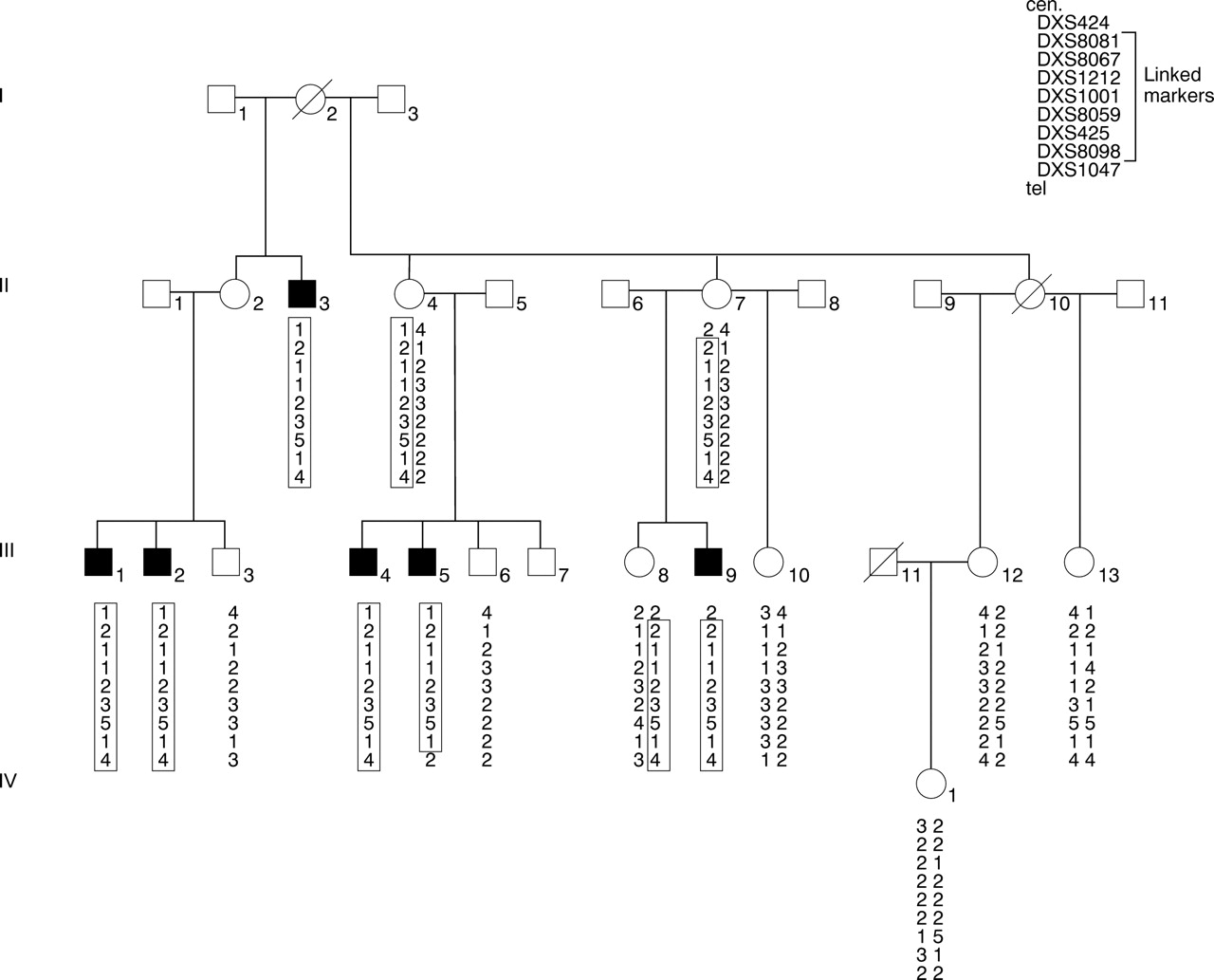
The pedigree (family K8135), with transmission of MR from five normal or minimally affected female carriers to seven affected males in three generations and the absence of male to male transmission, is consistent with X linked inheritance (fig 1). We examined five affected males and included information from the clinical records of a sixth affected member of the same family. No information is given on the sixth affected male (III.14), who declined to participate and is not included in the haplotype pedigree. Blood was collected for DNA studies from 15 family members after informed consent was obtained.

Partial pedigree of family K8135 showing haplotypes of microsatellite markers showing six affected males. A seventh affected male and sibs were omitted since blood samples could not be obtained. Boxes indicate haplotype inherited with disease phenotype.
DNA STUDIES
Genomic DNA was isolated from peripheral blood of members of family K8135 using a high salt precipitation method.3Specific dinucleotide or trinucleotide polymorphisms were generated as given in Nelson et al 4 or Dibet al. 5 Forward primers for the PCR primer pairs were synthesised and labelled with fluorescein amitide (FluorePrime, Pharmacia) on a Beckman 1000 DNA synthesiser and desalted through Sephadex G-25 (NAP-10 columns, Pharmacia). The polymorphisms were detected using an automated Laser Fluorescent Sequencer (ALF, Pharmacia) in conjunction with Fragment Manager (Pharmacia) and the software package, Automated Linkage Preprocessor (ALP).6CEPH person No 134702 was included in all microsatellite analyses as a means of accurately assigning the alleles at a particular locus.
LINKAGE ANALYSIS
Two point linkage analysis was done with 37 polymorphic markers spread along the entire X chromosome using the Fastlink 3.0 program.7 The gene frequency was set at 0.0001. Penetrance in males was set at 1.0.
Results
Patient III.1 was the most severely affected (fig 2). There was low birth weight (1900 g), recurrent apnoea, and incubator care for two weeks and these perinatal complications may have resulted in his more severe course. All developmental milestones were delayed. At the age of 18 months, he was admitted to hospital with pneumonia, convulsions, and otitis media. He has required anticonvulsant therapy since. His communication was primarily non-verbal but he was able to vocalise sounds. He seemed to understand commands. Examination at 38 years (fig2) showed an alert, non-distressed, and wheelchair bound person. Height was 157 cm (<3rd centile) but head circumference and eye measurements were normal. Head and neck examination showed normal hair implantation and a blind right eye (phthisis bulbi). The left eye was normal with a light reactive pupil that followed and tracked light. A prominent lower lip was present. Moderate cervicothoracic kyphosis was present. Cardiothoracic examination was normal. There was moderate truncal obesity. Genitourinary examination showed a normal penis and small testes (<10th centile). All joints were hyperextensible and there was muscle wasting of the legs, small feet, and a calcaneal valgus deformity. Reflexes, muscle tone, and sensation were within the normal range.
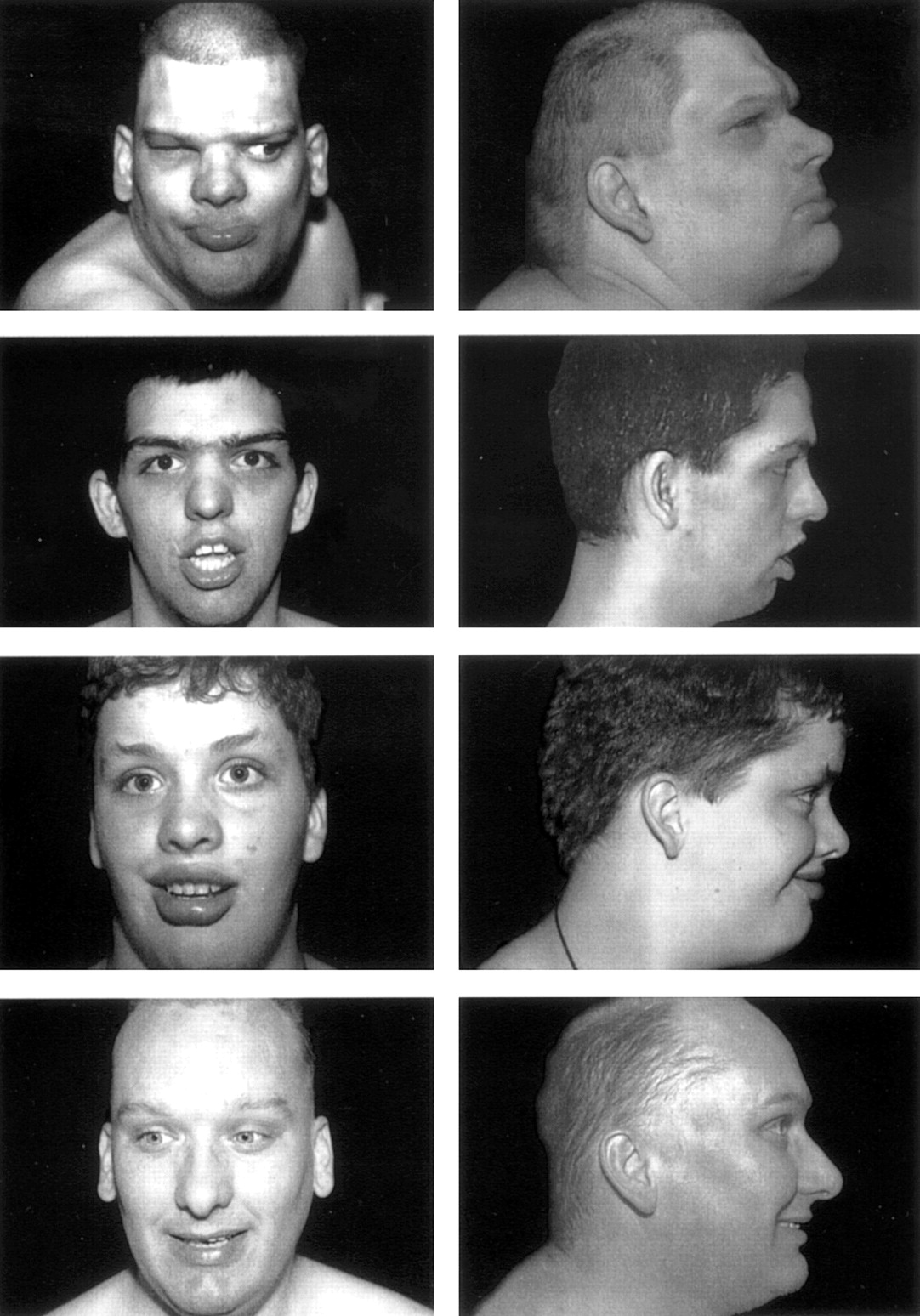
Facies of III.1, III.9, III.5, and III.2 (from top to bottom). Note prominent lower lips in all.
Patient III.2 was the least affected of the five males who were examined. No neonatal or childhood records were available except for a report of a functional systolic heart murmur detected at the age of 9 years. When examined at 40 years he was ambulatory and highly social, understood commands, and communicated verbally with good sentence structure. Height was 161 cm (<3rd centile). Weight, head circumference, canthal, and ear measurements were within normal values. There was baldness and some dental crowding, but his facial features were normal and a prominent lower lip was not present (fig 2). There was cervicothoracic kyphosis. Cardiothoracic and abdominal examination were normal except for mild truncal obesity. The penis was of normal length and testicular volume was 10 ml (<10th centile). Distal joints were hyperextensible. Hand measurements were normal and his feet were very small (size 5-6). Neurological examination showed mildly increased reflexes in all muscle groups and normal tone; there was muscle wasting of the legs. A fine tremor and decreased fine motor coordination in the upper limbs were present.
Patients II.3 (56 years old), III.4 (16 years old), III.5 (19 years old), and III.9 (21 years old) have most features in common with the above cases (table 1) except for the variation in severity of their mental retardation. Only III.1 had birth and neonatal complications. Tests for pituitary and endocrine function in III.9 were within normal limits and gave no evidence for panhypopituitarism or isolated growth hormone deficiency: FSH (6.0 mIU/ml), LH (5.0 mIU/ml), ACTH (26 pg/ml), IGF-1/somatomedin C (338 ng/ml), cortisol (7.6 μg/dl, serum), free thyroxine (0.8 ng/dl, serum), thyroxine (T4) (4.9 μg/dl, serum). It was not feasible to carry out a provocative test for growth hormone.
Clinical findings
Patient III.8 was the only carrier who was examined. Her medical history included normal weight, length, and head circumference. Developmental milestones were normal. She had been evaluated for Tourette syndrome and attention deficit disorder (ADD). It was reported that she had several tics (blinking, squinting, lip curling, shoulder shrugging), each present at different times, memory problems, and learning difficulties. Examination at 12 years showed only a thin habitus and mild tremor in her upper extremities.
CLINICAL COMMENTARY
The primary clinical findings are summarised in table 1. The ages of the affected males ranged from 16 to 50 years. Except for a prominent lower lip, the facies were normal and both stature and head circumference were normal at birth. The significant delay in growth and short stature found in five males was not documented until the prepubertal period. Testicular size was below the 3rd centile in the four affected adults who were examined. Although the testicular size in III.4 (aged 16) was not abnormal, the testicular volume (10 ml) was identical to the older affected males. If no further increase in testicular size occurs, then his testes will also be below the 3rd centile. The shoe sizes of affected adults varied between sizes 5 and 6. The gap between the first and second toes was striking in three of five affected males (two are shown in fig 3). Speech varied significantly in affected males. It was completely absent in one (III.1) and two (III.5 and III.9) had a vocabulary limited to only a few words. Only one (III.2) was able to communicate fairly well. Generally the affected males were passive but were easily frustrated and became aggressive when they were not able to communicate or did not receive enough attention. The tremor, present in five people, was present at all times and intensified during fine motor tasks. Endocrine studies in III.9 showed normal pituitary and other endocrine functions. Similarly, there was no evidence of isolated growth hormone deficiency in view of the normal IGF-1 in III.9.

Gap between first and second toes in III.5 and III.9.
LINKAGE RESULTS
Linkage analysis of 37 markers (table 2) resulted in a maximum lod score of 2.80 at DXS1212 and 2.76 at DXS425 with no recombination. The limiting markers were DXS424 (proximal) and DXS1047 (distal).
Family K8135: pairwise lod scores
Discussion
Consistent clinical findings in the affected males included short stature, obesity (fig 4), prominent lower lip, small testes and feet, hyperextensible joints, and kyphosis. In three of five subjects, a gap between the first and second toes was also found. Consistent neurological findings included fine tremor of the upper limbs, decreased fine motor coordination, significant muscle wasting of the lower legs in five subjects, and a wide based gait in three. Two were wheelchair bound. Decreased attention span, hyperactivity, and mood swings were present in 5/5 affected males. Aggressive behaviour was also present in 3/5 subjects. These clinical findings have not been previously reported in a family with XLMR.2
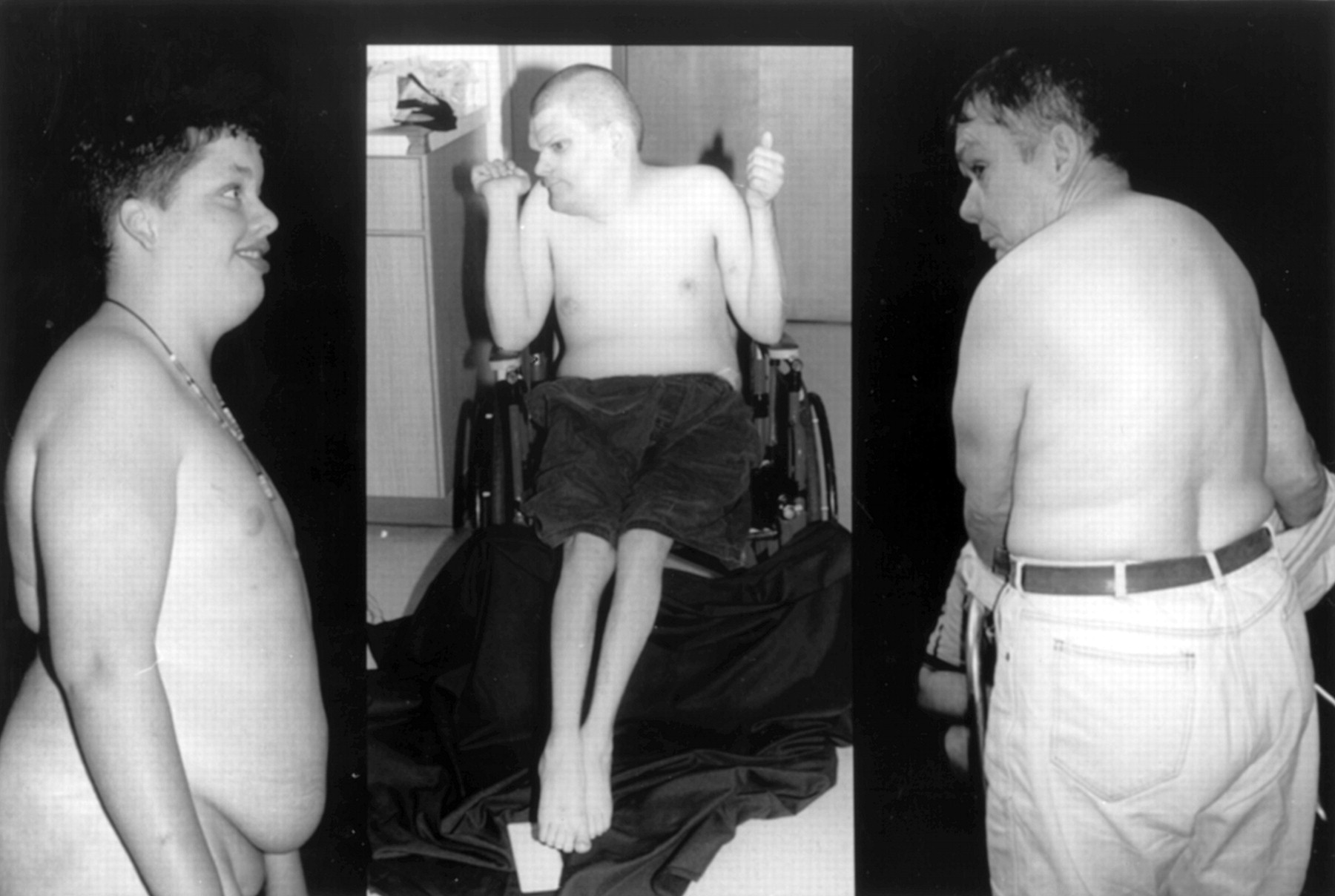
Obese habitus in III.5, III.1, and II.3 ( L to R).
The linkage results place the gene in Xq24-Xq25. As shown in fig 5, eight MRX families and 10 syndromes have localisations which overlap with the present family. The clinical differences make it unlikely these disorders are allelic, but allelism is possible for either the MRX or syndromic disorders since there has recently been a report of an MRX family resulting from a mutation in a known syndrome.8

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic of Xq24-Xq27.3 showing linkage limits for family K8135 and other XLMR entities that map to the region. DXS probe numbers are given at the limits of localisations.
The family reported by Turner et al 9 with mild Borjeson-Forssman-Lehmann syndrome (BFL) overlaps partially with the present localisation, that is, between DXS10 and DXS8098. In this family the majority of affected males had short stature, obesity, small testes, and a gap between the first and second toes. However, many significant differences were also present. No one in the present family had large ears or a small penis, and the BFL family had a different facies with no prominent lower lip or high palate. In addition, no one had small feet. The small toes in BFL were different and the muscle wasting and neurological findings shown in table 1 were not present. Although BFL and the present disorder might be allelic, on a clinical basis they appear different.
Thirty six of the known XLMR syndromes include short stature and hypogonadism occurs in 18. Panhypopituitarism (PHP) (MIM 312000)10 and isolated growth hormone disorder (IGHD) (MIM 300123)11 localise to the same region (fig 5). Both have short stature but neither syndrome includes small testes. Tests of pituitary and endocrine function in the present family also did not support either of these diagnosis. The tremor in the present family, which was a constant, fine tremor, differs from the Parkinsonian tremor seen in Waisman-Laxova syndrome (MIM 311510) and in the intention tremors described in the Pettigrew syndrome (MIM 304340) and ataxia dementia (MIM 301840).
The remaining XLMR syndromes (Christianson et al,12 Gustavson (MIM 309555), Arenaet al,13 Arts (MIM 301835), Christian (MIM 309620), Chudley et al,14 and Cowchock-Fishbeck (MIM 310200), which overlap the present localisation, do not have significant clinical similarities. Only molecular studies following the cloning of these several disorders will resolve the issue of possible allelism for this group of XLMR disorders. None of these disorders, however, has currently been cloned.
Acknowledgments
This study was funded in part by a National Institutes of Health grant (RO1 HD26202) to the University of Miami, School of Medicine (HAL) and the Greenwood Genetics Center (CES). The ethical and human aspects of these studies have been approved by Human Subjects Committees at each of the collaborating institutions.