Article Text

Abstract
The Smith-Lemli-Opitz syndrome (SLOS) is one of the archetypical multiple congenital malformation syndromes. The recent discovery of the biochemical cause of SLOS and the subsequent redefinition of SLOS as an inborn error of cholesterol metabolism have led to important new treatment possibilities for affected patients. Moreover, the recent recognition of the important role of cholesterol in vertebrate embryogenesis, especially with regard to the hedgehog embryonic signalling pathway and its effects on the expression of homeobox genes, has provided an explanation for the abnormal morphogenesis in the syndrome. The well known role of cholesterol in the formation of steroid hormones has also provided a possible explanation for the abnormal behavioural characteristics of SLOS.
- Smith-Lemli-Opitz syndrome
- cholesterol metabolism
- 7-dehydrocholesterol reductase
- clinical history
- management
Statistics from Altmetric.com
- Smith-Lemli-Opitz syndrome
- cholesterol metabolism
- 7-dehydrocholesterol reductase
- clinical history
- management
History
The Smith-Lemli-Opitz syndrome was first described in 1964 by the late David Smith, the Belgian paediatrician Luc Lemli, and John Opitz1 in a report of three patients who had in common a distinctive facial appearance, microcephaly, broad alveolar ridges, hypospadias, a characteristic dermatoglyphic pattern, severe feeding disorder, and global developmental delay. A more complete delineation of SLOS was presented in 1969 as the “RSH syndrome”, a non-descriptive acronym of the first letters of the original patients' surnames.2 The description of many new cases of SLOS over the next 20 years expanded the known characteristics of the syndrome, especially in the recognition of multiple internal anomalies (table1).2-11 Many affected children died in the first year from failure to thrive and infections, but many others survived to adulthood. Somewhat later, several authors described patients with a lethal syndrome that resembled SLOS, and which they designated “type II SLOS”.12-16 These children had many of the anomalies found in SLOS, but died in the newborn period from internal malformations. Moreover, most 46,XY “males” with so called type II SLOS had severe hypogenitalism or female appearing external genitalia. In all informative families, segregation of either form of SLOS was consistent with autosomal recessive inheritance.2 12 The genetic cause of the disorder was not suspected for many years, although by the mid 1980s a number of abnormalities of steroid metabolism in SLOS had been reported, including enlarged, lipid depleted adrenal glands17 and aberrant patterns of steroid sulphates in plasma and urine.12 17 18 Nevertheless the primary defect remained unknown until Natowicz and Evans19found that a patient with SLOS had essentially undetectable levels of normal urinary bile acids. An analysis of that patient's plasma sterols led to the discovery20 that the patient had a more than 1000-fold increase in the level of 7-dehydrocholesterol (cholesta-5,7-dien-3beta-ol; 7DHC), suggesting a deficiency of 7-dehydrocholesterol reductase (DHCR7), the final step in the Kandutsch-Russell cholesterol biosynthetic pathway.21 The same sterol pattern has subsequently been found in most patients with either type of SLOS, as well as in patients with variant syndromes that could not be assigned the diagnosis of SLOS on clinical grounds alone.22 23 Although initial evidence suggested that the human gene for SLOS was located at 7q32.1, the humanDHCR7 gene was later cloned and localised to chromosome 11q12-13 by Moebius et al.24 Shortly afterwards, three groups independently reported apparently disabling mutations ofDHCR7 in patients with SLOS.24-26
Findings in 167 clinically diagnosed cases of Smith-Lemli-Opitz syndrome compared with 164 biochemically confirmed cases
Clinical overview
GENERAL
Although most published reports of SLOS describe only a single patient or a small number of patients, there are a few exceptions in which 10 or more cases are described.2 9 12 23 27-29Furthermore, several excellent older reviews exist.10 15 30 A tabulation of the major clinical features of patients described in sufficient detail is provided in table 1. A comparison of the most common characteristics of patients ascertained by clinical v biochemical diagnosis shows similar frequencies of the physical anomalies. Only structural brain anomalies and anomalies of the genitalia are somewhat more common in the clinically diagnosed group. Before the recognition of the biochemical cause of SLOS, several authors suggested division of SLOS into the relatively less expressed type I form and the more severe type II form.12 13 31 Curry et al 12 cautiously suggested that type I and type II might differ genetically, whereas others thought the subdivision was not justified.32 To address this question, Bialeret al 15 devised a scoring system to evaluate the clinical severity of SLOS. Upon scoring 122 published cases, they found a unimodal frequency distribution of the scores for various anomalies, which was interpreted as evidence for a continuum of severity in SLOS and against a genetic distinction between types I and II SLOS. However, because length of survival was part of the original scoring in addition to separate scores for individual visceral anomalies, the original scoring system of Bialeret al 15 was overweighted for internal anomalies. Therefore, we (Kelley and Hennekam, unpublished data) modified the Bialer score, to weight embryologically separate organ systems equally. The revised scoring system (table 2) has been used in two series of biochemically confirmed cases23 27and showed a continuum of severity and strong correlations with various biochemical parameters. Subsequent molecular genetic studies confirmed that the differences in severity between types I and II SLOS are explained by the severity of the mutations responsible. However, these results do not preclude the existence of genetic heterogeneity in SLOS; recently two mildly affected sibs were found to have probably a related but biochemically different disorder of sterol metabolism.33
Adapted (from ref 15) severity score for anatomical abnormalities in Smith-Lemli-Opitz syndrome
INCIDENCE
The earliest estimate of the incidence of SLOS was made by Lowry and Yong,34 who found an incidence in British Columbia of 1/40 000 births and a carrier frequency of 1%. The incidence in that survey increased to 1/20 000 births when suspected but less definite cases were included. The incidence in a completely ascertained newborn population in Middle Bohemia (Czech Republic) was estimated to be greater than 1 in 10 000.35 However, in contrast to these relatively high incidences based exclusively on clinical diagnosis, the incidence of SLOS diagnosed biochemically in similar populations appears to be much lower. For example, between 1995 and 1998, when knowledge of the biochemical defect was widespread, the two laboratories that perform at least 80% of biochemical testing for SLOS in the United States identified only about 40 new cases per year, or an estimated incidence of less than 1 in 60 000 births (Kelley and Tint, unpublished data). Similarly, estimates of only 1 in 60 000 newborns in the United Kingdom,27 1 in 80 000-100 000 births in The Netherlands (Waterham et al, unpublished data), and an even lower incidence in Japan36 have been reported. The number of cases with African,23 37Asian,38 39 or South American40 ancestry is also low. Although there remains some uncertainty about the absolute incidence of SLOS in some countries, there clearly are strikingly different incidences among various ethnic groups. Both heterozygote advantage and founder effect have been suggested to explain the relatively higher incidence of SLOS among those of European descent. The percentage of patients born to consanguineous parents is relatively low for an autosomal recessive entity,23 27 35 which suggests persistence in the population of multiple mutant alleles through heterozygote advantage.
CRANIOFACIAL CHARACTERISTICS
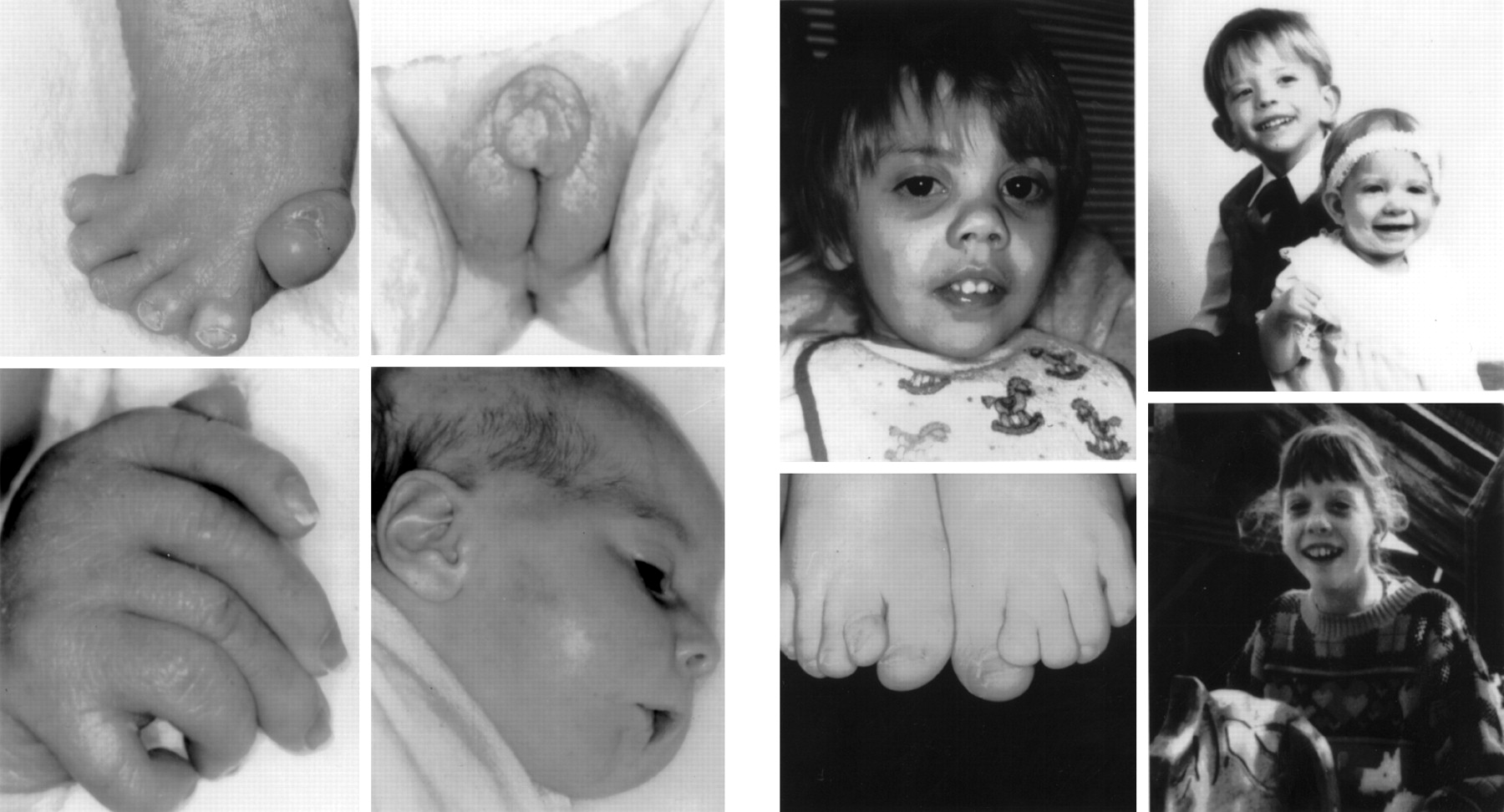
In the following listing of cases with various features, no indication is made of whether a case was biochemically confirmed or not. In general, cases published after 1994 will have been biochemically confirmed, but in cases from earlier papers this remains unknown. The “SLOS face” is highly characteristic of the syndrome and easily recognised in most patients, but may be very subtle in some.27 41 The most salient features are microcephaly, bitemporal narrowing, ptosis, a short nasal root, anteverted nares, and a small chin (fig 1).

Patients with Smith-Lemli-Opitz syndrome.
Congenital microcephaly is very common (for exact percentages of this and other symptoms see table 1). Although a prominent ridge along the metopic suture can often be palpated, true craniosynostosis is uncommon.2 12 More than half of the patients have ptosis, often asymmetrical or unilateral. The palpebral fissures are usually straight, but both upward and downward slanting occurs. Other less frequent external eye anomalies include hypertelorism, epicanthic folds, absence of lacrimal puncta,42 and unusually long cilia.43 Mild exophthalmos has not been reported often,2 but is relatively commonly seen in figures in publications. Ocular defects include mainly congenital and occasionally postnatal cataracts, strabismus, and nystagmus.11 44-46Vision is usually normal. Less common symptoms are sclerocornea,13 43 47 heterochromia iridis,48 coloboma of the iris,16 42posterior synechiae,6 45 49 glaucoma,2 12retinal hyperpigmentation,2 optic atrophy,2 12 45 optic pits,42microphthalmia,5 23 and abnormalities of eye movements.44 Aniridia was reported by Graciaet al,50 but the diagnosis in this mentally normal child with bilateral gonadoblastoma may be questioned.
A hallmark of the syndrome is the shape of the nose; usually the nasal bridge and base are broad, the nasal root short, and the nares anteverted. The nasal bridge can be flat or high, often with a striking capillary haemangioma extending across the glabella. The degree of anteversion of the nares decreases with age, but remains distinct in many adults.27 48 59 Narrow or atretic choanae may occur.51
The ears often appear low set and posteriorly rotated, but are otherwise unremarkable. Rarely, the ear canals have been reported to be very small.8 52 Congenital sensorineural hearing deficits may affect as many as 10% of patients, but many more severely affected children are not tested. Anomalies of the inner ear shown by radiography or found at necropsy have been described.53 54 The philtrum is long, as can be expected in shortening of the nose. Although cleft lip has been reported in SLOS,13 55-57 only the “midline” type of cleft associated with the holoprosencephaly sequence has been found in patients with abnormal cholesterol metabolism.58 In some patients, the mouth is large, which, combined with frequent micrognathia, gives a distinctive appearance.59 More severe micrognathia, including the classical Pierre Robin sequence, is not rare in SLOS.
The intraoral anomalies of SLOS are diagnostically important. The palate is usually highly arched, often with a midline cleft of the uvula, soft palate, or hard palate. In addition, the alveolar ridges typically are abnormally broad and conspicuously ridged. The tongue can be small with redundant sublingual tissue12 or sublingual cysts13 31 in the more severely affected children. More rarely, the tongue is bifid.27 Crowded teeth and widely spaced incisors are not uncommon,27 44 52 and oligodontia or polydontia,44 unusually large central upper incisors,27 59 enamel hypoplasia,10 and premature tooth eruption60 have been reported. A detailed dental study in SLOS, however, has not been published. Pharyngeal abnormalities have included a small larynx13 and small vocal cords with a subglottal shelf of excess fibrocartilaginous tissue.12
The neck can appear short and excessive skin folds or nuchal oedema are common, especially prenatally.61
CENTRAL NERVOUS SYSTEM
The structural brain abnormalities of SLOS have been reviewed by Garcia et al,62 Cherstvoyet al,63 and Marionet al.64 In addition to microcephaly, which is almost universal in SLOS, common abnormalities include enlarged ventricles,2 12 52 63 65 66hypoplastic or absent corpus callosum,12 27 57 63 66 67 hypoplastic frontal lobes,3 9 and pituitary lipoma.68 Cerebellar hypoplasia, sometimes with severe hypoplasia or aplasia of the vermis, is also not uncommon.2 5 9 27 44 52 53 62 64 67Various forms of the holoprosencephaly sequence occur in about 5% of patients.5 13 17 23 58 On histological examination of the brain, the most important findings are disturbed cerebral neuronal migration,12 27 64 67 69 72 73 extensive gliosis,12 dysplasia of the medial olivary nuclei, and ectopic Purkinje cells.9 44 Several authors27 44 62 67 70-72 74 have reported maturational abnormalities of the white matter; however, most cranial MRI studies do not show white matter abnormalities. Although seizures are not uncommonly reported in SLOS, including infantile spasms,32 75 they are uncommon in biochemically proven cases of SLOS and may not be substantially more frequent than in children without SLOS.2 3 7 27 48 52 62 69 76 The same reservation must be applied to a number of central nervous system malformations reported in older SLOS publications before biochemical confirmation was available.
SKELETAL ANOMALIES
The skeletal anomalies have been reviewed in detail.10 15 23 27 63 Bilateral or unilateral postaxial polydactyly can be present in the hands or, less commonly, the feet, or both. Preaxial polydactyly has not been reported in a biochemically proven case. The thumb is generally short and proximally placed and the first metacarpals and thenar eminences are typically hypoplastic.2 12 77-79 Other unusual digital abnormalities include ectrodactyly,56 70 80monodactyly,80 oligodactyly,56 81 82 radial agenesis,80 brachydactyly, absent middle phalanx of the second finger, radial or ulnar deviation of the fingers, clinodactyly, camptodactyly, and various syndactylies.2 10 12 27 56 70 80 Rhizomelic and mesomelic limb shortness and, more rarely, “chondrodysplasia punctata” occur in SLOS, but a true chondrodystrophy is not found.12 13 16 23 27 29 63 70 Dermatoglyphics in SLOS have been reported to be distinctive with an increased proportion of whorls and decreased proportion of ulnar loops on the finger tips in most series,2 10 12 52 but not all.83 The presence of whorls may point to a pathogenesis through puffy finger tip pads or oedema during early fetal development. There is no study as yet of dermatoglyphic findings in a group of biochemically proven cases, but whorls appear to predominate as commonly reported in the early case reports.
One of the most consistently present anomalies in SLOS is the distinctive “Y shaped” cutaneous syndactyly of the second and third toes, which has been reported in up to 99% of biochemically proven cases.23 27 Postaxial polydactyly of the feet is common in severe SLOS, and sometimes takes the form of polysyndactyly with a “windswept”” foot deformity.12 13 15 16 27 45 48 63 72 84 Other lower limb abnormalities include club foot, varus or valgus foot deformities, short first toes, and hip dislocations.9 12 27 31 Occasionally reported skeletal abnormalities include dense base of the skull,31scoliosis,34 52 69 78 kyphosis,35 71 78ovoid vertebrae,68 cervical ribs,54 78 82thin ribs,57 68 85 and missing ribs.71Although epiphyseal stippling (“chondrodysplasia punctata”) has been reported in a few cases,1 32 57 68 86 such stippling has been found in only one biochemically confirmed patient,87 who also had a de novo balanced chromosome translocation.
GENITAL ANOMALIES
The genitalia in male SLOS patients range from normal to the appearance of complete sex reversal.12-15 60 65Classically, hypospadias varies from coronal to perineoscrotal hypospadias, although the latter is uncommon except in the biochemically most severely affected cases. Maldescent of the testes is common, but, even with severely malformed genitalia, the testes are often easily palpated in the scrotum, which is sometimes bifid.72 88 Mullerian duct derivatives are usually absent in 46,XY males, as expected, but blind ending vagina, rudimentary or bicornuate uterus, and persistent cloaca have been described.12 13 15 31 51 89 The gonads vary from normal testes to ovotestes to normal ovaries, or may be missing.5 In females, the external genitalia may appear normal or there may be distinct hypoplasia of the labia majora and minora. There are also single reports of premature thelarche and high serum prolactin levels in a 15 month old girl with SLOS,90and a malignant germ cell tumour with a contralateral streak gonad in another female.91 Menstrual function is often irregular but otherwise normal in most SLOS adolescent females and adults, although menarche is often delayed. One adolescent girl with (biochemically unproven) SLOS and borderline intelligence gave birth to an apparently normal daughter.34
CARDIOVASCULAR ANOMALIES
The cardiac anomalies of SLOS have been reviewed by Robinsonet al,7 Johnson,10and, most extensively, by Lin et al.92 Almost half of SLOS patients have a congenital heart defect, although if only biochemically confirmed patients are taken into consideration, this percentage is somewhat lower.23 27 92 There is a strong predominance of endocardial cushion defects and the hypoplastic left heart sequence, whereas conotruncal defects are uncommon. Almost every known cardiac defect has been described at least once. The five most prevalent defects found in a study of 95 biochemically confirmed cases of SLOS were atrioventricular canal (25%), primum atrial septal defect (20%), patent ductus arteriosus at term (18%), and membranous ventricular septal defect (10%).92 Lin et al 92 hypothesised that the abnormal development of the extracellular matrix may be the cause of both the cardiac defects and the absence of ganglion cells in the bowel (Hirschsprung disease) because of altered cell membranes and cell to cell interactions. However, abnormal migration or proliferation of neural crest derived cells, which contributes substantially to the endocardial cushions, could also explain both the cardiac defects and abnormal intestinal ganglion cells. In addition to structural heart defects, there is a substantially increased frequency of pulmonary hypertension in the newborn period and persistent hypertension postnatally, but limited largely to patients with especially low cholesterol levels, possibly related to the abnormal steroid metabolism of these patients.69 71 72 93
RENAL AND ADRENAL ANOMALIES
About one quarter of patients with biochemically confirmed SLOS have renal anomalies,13 27 94 most commonly renal hypoplasia or aplasia,10 12 13 27 55 94 95 renal cortical cysts,9 12 27 44 57hydronephrosis,9 10 27 44 60 67 72 renal ectopia,1 9-31 44 57 67 72 79 ureteral duplication,31 60 and persistent fetal lobation.15 27 31 79 A number of cases with the oligohydramnios sequence caused by bilateral renal aplasia or other renal causes of severely diminished urinary output have been described.12 13 55 82 95 The bladder and ureters may be hypoplastic, probably secondary to renal hypoplasia or aplasia.
Both adrenal hyperplasia12 17 and adrenal hypoplasia13 have been reported in SLOS, but the growth and shape of the adrenals appear to be normal in most.13 29 Histological studies of hyperplastic SLOS adrenal glands17 typically show deficient cortical lipid, where normally fetal adrenals contain much cholesterol. Postnatal studies of adrenal function in children with SLOS have shown either normal function73 or, in several biochemically severely affected children, decreased steroid synthesis.96
PULMONARY ANOMALIES
Abnormal pulmonary lobation and pulmonary hypoplasia are common in the more severely affected cases of SLOS.9 12 13 31 51 53 82 As expected, pulmonary hypoplasia is also common in SLOS patients with the oligohydramnios sequence secondary to renal aplasia.12 13 55 82Accessory pulmonary arteries have also been described.45Anomalies of the laryngeal and tracheal cartilages are common even among patients with mild forms of SLOS and may cause obstructive sleep apnoea. Serious complications have resulted from difficulties with emergent and even elective intubation because of marked tracheal narrowing and other abnormalities of the laryngeal and tracheal structures.69
GASTROINTESTINAL ANOMALIES
Pyloric stenosis is a prominent clinical problem noted in the original description of SLOS and in many subsequent case reports.1 2 7 97 Pyloric stenosis in SLOS has the same clinical and anatomical characteristics as in otherwise normal children, but vomiting and other feeding problems commonly persist after surgical repair, in part because of apparent intrinsic abnormalities of intestinal motility. In the more severe cases, intestinal aganglionosis occurs, both short segment and more extensive involvement of the upper and lower intestinal tract.12 87 98 99 Even among SLOS patients lacking histological evidence of intestinal aganglionosis, intestinal dysmotility is common, especially in the first year. However, whereas pyloric stenosis historically has been reported in at least 10% of SLOS patients, it is now uncommon in SLOS patients treated with supplementary cholesterol starting shortly after birth (Kelley, unpublished data).
A small number of SLOS patients have had the unusual finding of dysplasia or aplasia of the gall bladder12 27 or gallstones in infancy or later childhood.12 69 More common, however, is transient or, more rarely, lethal cholestatic liver disease.12 27 67 100 Histologically, iron pigment in liver cells has been found to be increased,51 67 as is also seen in peroxisomal disorders. Although lipidosis is not commonly associated with SLOS, diffuse lipid storage was reported by Parneset al, 74 and Porteret al 101 recently described impaired intracellular trafficking of LDL in SLOS fibroblasts.
Pancreatic islet cell hyperplasia has been reported frequently in severe SLOS.9 12 17 31 65 The histology in these cases features nesidioblastosis and reduced quantities of somatostatin.17 102 Other abdominal malformations less frequently reported include intestinal malrotations,9 12 27 45 84 absence of the diaphragm or diaphragmatic hernia,9 55 polysplenia and asplenia,9 anal stenosis or atresia,9 13 52 65 103 and Meckel's diverticulum.2 7
OTHER ANOMALIES AND CLINICAL PROBLEMS
Involuted67 and hypoplastic99 thymus and absent parathyroids12 have been found. Among the more common dermatological and hair abnormalities are hypopigmented hair,2 78 mild to extreme skin photosensitivity in more than half of patients,27 hyperhidrosis of the palms,104 marked cutis marmorata,99 and eczema.27 After infancy, many children have mild to marked acrocyanosis that appears to be autonomic in nature and often resolves after cholesterol therapy. Widely spaced nipples are mentioned repeatedly, but measurements are rarely provided. There are two reports of excessive muscle rigidity after halothane anaesthesia, but diagnostically raised creatine kinase levels were not found.105 106
Natural history
The neonatal period and infancy of SLOS patients are almost invariably disturbed by feeding problems, including poor or abnormal suck, swallowing difficulties, vomiting, and lack of interest in feeds. Oral tactile defensiveness and failure of progression to textured foods in later months is also characteristic of the SLOS infant. As a result, more than 50% of patients require nasogastric tube feedings, often progressing to gastrostomy feeding for several years. As expected, patients with a cleft palate and a small mandible (Pierre Robin sequence) have the most severe feeding problems. Although gastro-oesophageal reflux is a common problem in patients with SLOS, such reflux is often caused by a failure to recognise that the children have congenitally small stomachs and intestinal dysmotility. In addition, gastro-oesophageal reflux secondary to milk or soy protein allergy seems to be unusually common in children with SLOS. Pyloric stenosis is also frequent in SLOS as shown by Linet al,92 who found that pyloric stenosis occurred in at least 7% of their biochemically confirmed cases and 11% of published cases. With early recognition of these causes of gastro-oesophageal reflux and vomiting in SLOS, fundoplications and other surgical interventions can be minimised.
Severe hypotonia is almost universal in SLOS during infancy. Although the hypotonia is partly central in origin, congenital muscle hypoplasia also contributes to the hypotonia. However, during the second year, muscle mass and tone often improve, and muscle strength and tone are typically normal in older children with SLOS. In later childhood, increased muscle tone may occur and can lead to joint and skeletal problems in non-ambulatory children. Such hypertonia appears to be extrapyramidal rather than spastic.
“Failure to thrive” is also almost universally diagnosed in children with SLOS, but the diagnosis more often than not is incorrect. Infants with SLOS are small for gestational age and most continue to grow below the 3rd centile despite adequate caloric intake, indicating a fundamental, genetic hypotrophy as the basis of the growth retardation. Weight gain can be even poorer in the first two years because of feeding and GI motility problems. Although even well nourished infants will experience a fall off from their birth centiles over the first year, most children with SLOS have normal growth velocities for length and weight in later years. Similarly, although head circumference is proportionately the smallest measurement at birth and postnatally, head circumference also usually remains proportionate with other measurements. Most measurements for classical SLOS fall between −1 and −5 SD below normal, but measurements as low as −8 to −10 SD occur in the more severely affected patients. With a few exceptions at both extremes, final adult height and head circumference are between 2 and 5 SD below normal.12 13 27 107 In several published series, final height in adults with “type I” SLOS was between 143 and 170 cm.27 48 59 Size at birth and growth of the biochemically more severely affected patients is substantially less.
During both infancy and childhood, children with SLOS appear to have an increased number of infections. Although many of the infections are otitis media, skin infections and pneumonias also seem to occur more often. Despite the frequency of reflux and hypotonia in children with SLOS, aspiration pneumonia is surprisingly uncommon, most probably because of the children's exaggerated gag reflex. Except for a single report of abnormal monocyte oxidative metabolism,108 no specific primary immune disturbances have been described in SLOS. However, death from sudden overwhelming infections is not rare in SLOS and suggests a fundamental abnormality in immune defenses or, possibly, adrenal function.96 Apart from the high frequency of milk and soya protein allergy71 and, possibly, an increased frequency of reactive airway disease, other primary immunological diseases do not seem to be common in SLOS.
MENTAL DEVELOPMENT AND BEHAVIOUR
With rare exceptions, global psychomotor retardation is characteristic of SLOS. Although, historically, most patients have been described as severely mentally retarded (IQ 20 to 40), such apparently poor development in part reflected difficulties in testing. In general, SLOS children are very sociable, have much better receptive than expressive language, and may be surprisingly mechanically adept for their apparent degree of cognitive impairment. Because of their poor expressive language and hyperactivity, routine developmental testing often underestimates their cognitive abilities. Gross motor development is typically more severely delayed than fine motor development, but most children with classical SLOS learn to walk between 2 and 4 years. With the availability of biochemical testing and the more frequent recognition of more mildly affected SLOS children, the known developmental spectrum of SLOS has widened substantially. Approximately 10% of children with biochemically diagnosed SLOS have development in the mildly retarded range (IQ 50 to 70). A few patients with normal or borderline normal development have been described,27 34and it is likely that the proportion of recognised patients with borderline and normal intelligence will increase.
In the newborn period, excessive sleeping and poor responsiveness are common. However, hours of shrill screeching or inconsolable screaming, especially at night and in the early morning hours, is a major behavioural characteristic of untreated SLOS later in infancy.10 27 Others appear hypersensitive to all visual and auditory stimuli and must be kept in quiet, dark rooms. Ryanet al 27 drew attention to the strikingly diminished amount of sleep in early childhood, which they found in 70% of their patients, some of whom slept for only two or three hours at night. Although this abnormal sleeping pattern may improve with age, adult SLOS patients with similar sleep problems are known. Many patients, even those with very mild clinical disease, may show self-injurious and aggressive behaviour. Most characteristic among these behaviours over the age of 3 years are forceful hyperarching (what we have called “opisthokinesis”), with or without head banging, and arm and hand biting. Marked tactile hypersensitivity of the hands and feet is also seen in more than 50% of patients. Behavioural characteristics of autism, such as hand flapping, abnormal obsessions, insistence on routine, and poor visual contact, are common in children with SLOS. Despite these many behavioural problems, most parents describe their children often as loving, affectionate, and happy.2 27
LIFE EXPECTANCY
Some investigators have suggested that there is an increased rate of spontaneous abortion in families with SLOS, but Ryanet al,27 in the only systematic study of prenatal losses, found 39 probands, 51 healthy sibs, 16 spontaneous abortions, and seven elective terminations among 43 sibships with 113 known conceptions. Furthermore, Kratz and Kelley95 found no increased recognised miscarriage rate and an expected segregation ratio of 25% in a cohort of prospectively monitored pregnancies. Although these data do not support the hypothesis of an increased miscarriage rate,10 109 110it may still occur in families with more severely affected children, since severe cardiac or renal abnormalities have been known to lead to mid-trimester intrauterine death of SLOS fetuses. As supported by the data of Cunniff et al, the low 17% segregation ratio found in one large series10 could be explained by the inclusion of a proportion of genetically different disorders at a time when biochemical confirmation of the diagnosis was not possible.
There are no recent figures for life expectancy in SLOS. Johnson10 found that 27% of cases in her series died before 2 years of age. However, an analysis of the causes of death was not provided. As ascertainment of SLOS, even in the era of biochemical diagnosis, has been incomplete, the exact percentage of cases with early lethality remains uncertain. Clearly, however, life expectancy in SLOS is determined largely by the severity of the internal malformations and the quality of general supportive care and not by an intrinsic degenerative process or biochemical toxicity per se.
Differential diagnosis
Many papers describe patients with a clinical phenotype resembling SLOS, especially before the era of biochemical diagnosis. In German publications the designations “Ullrich-Feichtiger syndrome”111 or “Typus Rostockiensis”,112 a multiple congenital anomalies syndrome with facial anomalies, polydactyly, and hypospadias, probably describe severe forms of SLOS.113 Among clinical diagnoses that have been proven or suspected to be cases of SLOS are the acrodysgenital syndrome,31 85 114Gardner-Silengo-Wachtel syndrome (OMIM 231060),115 116and holoprosencephaly-polydactyly (“pseudotrisomy 13”) syndrome (OMIM 264480).117 118 However, the predominance in the Gardner-Silengo-Wachtel syndrome of conotruncal malformations and in holoprosencephaly-polydactyly syndrome of gonadal dysgenesis, both uncommon abnormalities in SLOS, suggests that some of these biochemically untested patients do not have SLOS. Among diagnoses that have been incorrectly assigned to patients with SLOS are Noonan syndrome (OMIM 163950), Opitz syndrome (OMIM 145410, 300000), and Zellweger syndrome (OMIM 214100). Conversely, several patients with α thalassaemia-mental retardation syndrome (OMIM 301040) have been given the diagnosis of SLOS. Other disorders that resemble SLOS, but less so, are Meckel syndrome (OMIM 249000), hydrolethalus syndrome (OMIM 236680), Pallister-Hall syndrome (OMIM 146510), orofaciodigital syndrome type VI (OMIM 277170),12 13 112 118 119 and a number of individual case reports.74 120-124
Sterol biosynthesis
Cholesterol is a 27-carbon, mono-unsaturated sterol, synthesised from lanosterol by a series of oxidations, reductions, and demethylations, mostly limited to the endoplasmic reticulum (fig 2). The markedly increased levels of 7DHC with almost no 7-dehydrodesmosterol in patients with SLOS suggests that the principal route of cholesterol biosynthesis in man may be the Kandutsch-Russell pathway.21 Nevertheless, the relative abundance of desmosterol in neuronal tissues, the testes, and breast milk125-127 suggests that desmosterol may be the penultimate sterol in some tissues, or have specific functions in the tissue where it is abundant.
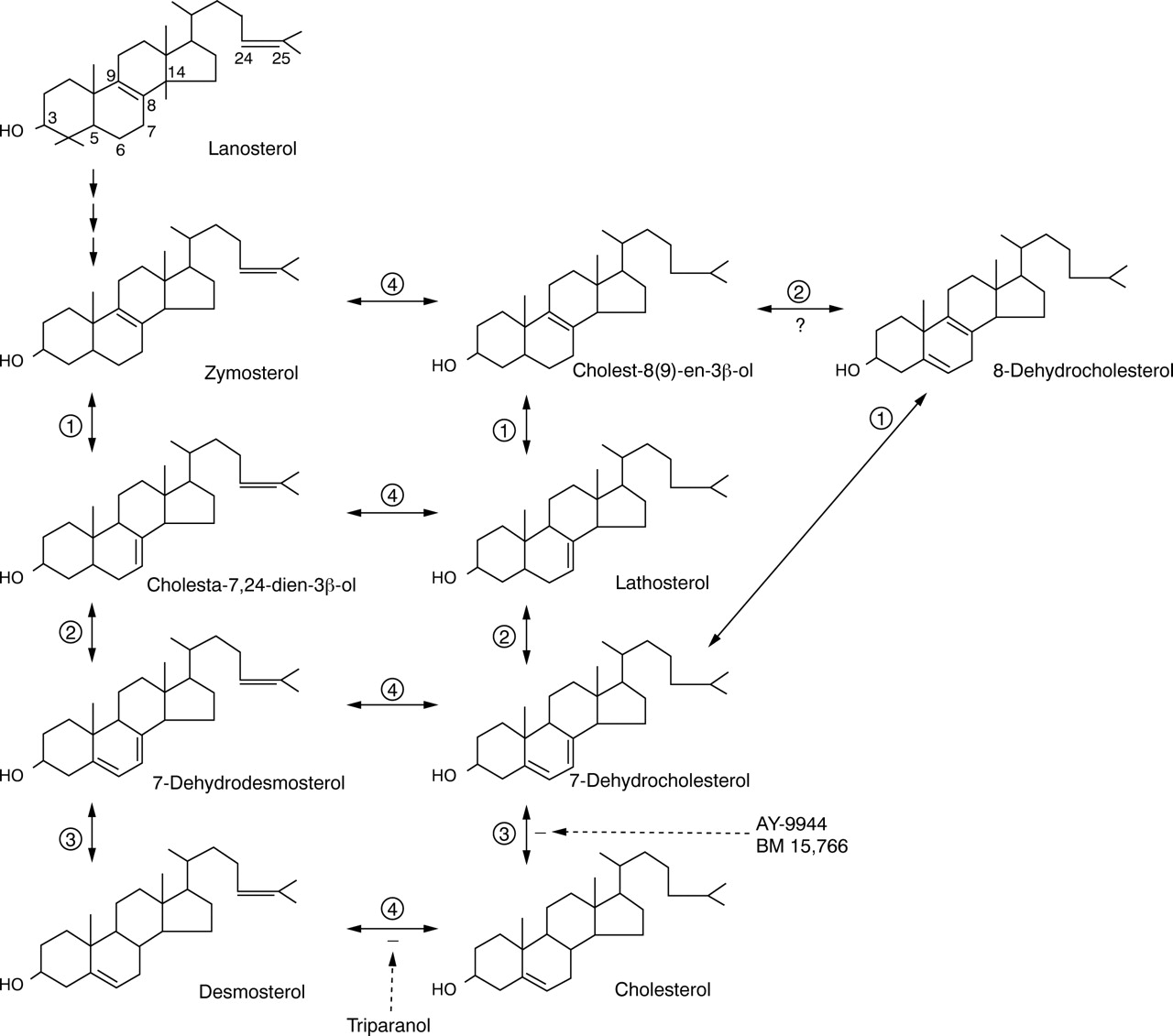
Enzymatic steps and major sterol intermediates comprising the pathway of cholesterol from the first sterol, lanosterol. The denoted enzymatic steps are (1) 3beta-hydroxysteroid-delta8,delta7-isomerase (EC 5.3.3.5); (2) 3beta-hydroxysteroid-delta5-desaturase (lathosterol dehydrogenase, EC1.3.3.2); (3) 3beta-hydroxysteroid-delta7-reductase (DHCR7,EC1.3.1.21); and (4) 3beta-hydroxysteroid-delta24-reductase (desmosterol reductase).
Before discovery as the marker metabolite of SLOS, 7DHC was known as a minor constituent of plasma and solid tissues. It was found to be increased up to three-fold in conditions associated with increased loss of bile acids, for example, ileal resection, and, therefore, increased cholesterol synthesis. 7DHC has special physiological importance as the precursor of vitamin D3 via photic conversion of 7DHC to pre-vitamin D3 in skin.
Whereas most of the early steps of cholesterol biosynthesis are well characterised at the DNA, RNA, and protein levels, the individual enzymes, cofactors, carrier proteins, and intracellular transport steps involved in the conversion of lanosterol to cholesterol are less well understood, as is the complex intracellular trafficking of cholesterol. A genetic disruption of one of these transport pathways appears to be the cause of the lipid storage in type C Niemann-Pick disease.128 Understanding intracellular trafficking and regulation of cholesterol uptake as well as de novo cholesterol synthesis will be important to understanding the cellular pathology of SLOS.
In contrast to many other small metabolites, very little cholesterol is transported after the first trimester from the mother to the fetus. Instead, most cholesterol must be synthesised by the fetus.129 130 Both in later fetal life and after birth, most if not all cholesterol in the brain is synthesised locally, not transported from blood lipoproteins.130-133 However, recent molecular biological studies have delineated a system for the delivery of maternal LDL to LDL receptors in the embryonic neuroepithelium in the first trimester.134 135 The fundamental importance of this system is supported by the discovery135 that transgenic mice lacking megalin, the LDL receptor in the embryonic neuroepithelium, develop holoprosencephaly, which has been linked to abnormal cholesterol metabolism at several levels.58 136 137 Thus, whereas delivery of cholesterol to the fetus after development of the placenta may be minimal, critical aspects of embryonic tissue differentiation may be sensitive to maternal blood cholesterol and LDL levels.
Although most cholesterol in tissues serves as a major structural lipid of membranes, some cholesterol also enters pathways for bile acid metabolism and the synthesis of steroid hormones. Another unexpected and recently discovered role of cholesterol is as a covalently bound element of active hedgehog proteins, a family of embryonic signalling proteins.58 137 138
Already in 1966, Roux and Aubry139 had shown that inhibitors of enzymes of the distal cholesterol biosynthetic pathway caused holoprosencephaly, microcephaly, pituitary agenesis, limb defects, and genital anomalies in the pups of exposed pregnant rats or mice. The same group also showed that feeding cholesterol to the pregnant, treated rats substantially limited or blocked the teratogenic effects of AY-9944, an inhibitor of DHCR7.140 Before 1993, holoprosencephaly had been reported in only one SLOS patient.17 However, using an increased 7DHC level as a diagnostic marker for SLOS, holoprosencephaly of variable severity has been found to occur in about 5% of patients with SLOS, most of whom have also been found to have mutations inDHCR7.23 58 This association was soon complemented by the discovery at about the same time138 that targeted disruption in mice of Sonic hedgehog (Shh) causes not only holoprosencephaly, but also distal limb defects and other skeletal anomalies. The same group showed that covalent addition of cholesterol to the equivalent hedgehog protein inDrosophila, hh, was an essential part of the “autoprocessing” of hh,137 141 wherein precursor Shh protein in the presence of cholesterol cleaves itself into a non-signalling COOH-terminal half and a mature, cholesterol substituted, N-terminal half, “hh-N”. The processed amino half appeared to possess all hh signalling activity, which in vertebrates includes patterning of development in the ventral forebrain and limb buds. The hh-N fragment appears to have a role in the attachment and localisation of hh-N to cell membranes.137 The proteins in vertebrates homologous to Shh, Desert hedgehog and Indian hedgehog, appear to have similar roles in, respectively, genital and skeletal development, both important aspects of malformation in SLOS.142 143 Another link between these malformations and hedgehog proteins was made by Roessler et al,136 who found that heterozygosity for mutations in SHH, the 7q36 linked gene encoding SHH in humans, causes autosomal dominant holoprosencephaly.
This convergence of holoprosencephaly, SLOS, Shh, and cholesterol metabolism focused attention on the possibility that the covalent attachment of cholesterol to Shh-N and related hedgehog proteins is the link between the abnormal cholesterol metabolism of SLOS and abnormal morphogenesis in SLOS. However, Cooper et al 144 showed that autoprocessing of hedgehog is not impaired when cholesterol in the reaction medium is replaced with 7DHC or any of many other 27-carbon sterols. In vitro studies with AY-9944 and other teratogens that cause SLOS-like malformations in rats strongly suggested that the defect in Shh signalling resides in sterol mediated changes in the target tissue and not an abnormality of the Shh-N signal itself.144 There are other possible levels at which the sterol defect of SLOS may cause impaired signalling of Shh, such as the sterol sensing domain in Patched,145 and the lamin B receptor, which has both sterol-14-reductase and a binding site structure similar to that of DHCR7.24 146 Shh is just one of several similarly functioning hedgehog proteins, all of which may interact with Patched in a sterol sensitive manner. Thus, it is possible that impaired signalling activities of Desert hedgehog142 and Indian hedgehog143 also play a role in SLOS in the abnormal morphogenesis of tissues that are not special targets of Shh signalling.
Other less specific disturbances may also contribute to the abnormal morphogenesis of SLOS. Cholesterol is severely deficient in all tissues, and such a severe disturbance in membrane sterol composition may affect developmental processes that involve cell-cell interactions, as suggested by DeHart et al.147 Interestingly, in recent studies with the SLOS mimicking teratogen BM 15 766, Lanoue et al 148 showed that more severe and more characteristic SLOS malformations were produced when BM 15 766 was given to mice with a genetic deficiency of LDL receptors, suggesting that, in embryonic tissue, a deficiency of cholesterol itself does indeed have a contributory role in the abnormal embryologic signalling of SLOS.
Despite the apparent negligible transfer of cholesterol from the mother to the human fetus, the most severely affected SLOS patients have measurable cholesterol levels at birth, typically 5 to 20 mg/dl.23 131 Moreover, initial rates of cholesterol synthesis in cultured fibroblasts from patients predicted on the basis of their mutations to have no DHCR7 activity may be as high as 50% of all sterols (Kelley, unpublished observations). Thus, there may be another genetic source of DHCR7 activity or a pathway of cholesterol synthesis not requiring DHCR7. The most likely source of such activity is the peroxisome, which has been shown to have an essential role in the early steps of cholesterol biosynthesis.149-151
Clinical severity correlates best with the level of cholesterol (inversely) or with 7DHC (or the sum of all dehydrosterols) expressed as a fraction of total sterols.23 The dehydrosterol fraction better expresses the systemic sterol abnormality than absolute blood sterol levels, which are subject to wide physiological variations independent of the rate of sterol synthesis. SLOS patients with multiple internal anomalies and early death have had, in general, much lower total sterol levels and higher 7DHC/sterol ratios than surviving SLOS patients.23 152 Although it was suggested153 that essentially all SLOS patients with cholesterol levels lower than 10 mg/dl die at birth or in the first few months,152 death in SLOS is caused largely by specific visceral malformations rather than some biochemical consequences of the abnormal sterol levels. Patients with levels less than 10 mg/dl at diagnosis who are long survivors are not rare and are usually distinguished by a lack of major internal anomalies. The genetic basis of the differences in biochemical and clinical severity is supported by the finding that sibs with SLOS usually have similar plasma levels of 7DHC and cholesterol and similar degrees of clinical severity.23 Indeed, subsequent studies ofDHCR7 mutations have shown a strong correlation between the predicted enzymatic effect of a patient's particular mutations and the clinical phenotype.163
As expected for an autosomal recessive disorder, the parents of SLOS patients have no or minimal physiological signs of abnormal sterol metabolism. About half of the parents are found to have mildly increased plasma 7DHC levels, with a mean level for all parents that is slightly higher than normal.153 However, more than 90% of SLOS parents have abnormally increased 7DHC to cholesterol ratios when their lymphoblasts are cultured in cholesterol depleted medium.33 Although there are no obvious adverse clinical consequences of mildly abnormal 7DHC metabolism in heterozygotes, no systematic survey for associated minor anomalies has been performed.
Apart from occasional case studies reporting adrenal or gonadal steroid levels,17 72 95 154 there have been few studies of steroid metabolism in SLOS. Reports of DHEA levels have shown both abnormally low and high levels, and testosterone levels were found to be either low or normal, with normal to mildly increased levels of FSH and LH in some. However, the anecdotal and usually uncontrolled nature of these studies precludes an informed understanding of sex steroid metabolism in SLOS. Moreover, the high frequency of hypogenitalism in SLOS is not sufficient evidence to implicate inadequate steroid production in utero, as the persistence of Mullerian remnants in some SLOS patients15 suggests defective genital morphogenesis unrelated to steroid hormone levels, possibly mediated through sterol related dysfunction of Desert hedgehog in genital tissues.155
Molecular genetics
The finding of hypocholesterolaemia and markedly increased levels of 7DHC in patients with SLOS immediately focused attention on 7-dehydrocholesterol reductase (DHCR7, EC 1.3.1.21) as a candidate deficient enzyme causing the disorder. Additional evidence came from drugs that inhibit distal steps of cholesterol biosynthesis produced SLOS-like malformations in mice and rats, suggesting that the cholesterol lowering action of these drugs may rather be the principal teratogenic factor.147 156-158 Before the renewed interest in DHCR7 caused by SLOS, DHCR7 had been little studied at a structural or DNA level, and then mostly in lower organisms.159 At the time of the discovery of apparent DHCR7 deficiency in SLOS, the chromosomal location ofDHCR7 was unknown, but 7q32.1 was thought to be the best candidate, because two unrelated SLOS patients were known to have de novo balanced translocations at that position. Further genetic study of one of the biochemically confirmed translocation patients160 showed that the 7q32.1 breakpoint interrupted not a gene involved in sterol metabolism but the human metabotropic glutamate receptor 8 (GRM8), which is not known to have a role in cholesterol biosynthesis. However, Moebius et al 24 shortly afterwards reported the cloning and mapping of a human microsomalDHCR7 gene to chromosomal location 11q12-13. Interestingly, DHCR7 was first cloned not as a gene for a sterol metabolising enzyme, but as a candidate gene for a “sigma type” drug binding protein.161 Only after complete sequencing was the gene found to have strong homology withDHCR7 from Arabidopsis thaliana. Within months, three groups identified disabling mutations in DHCR7 in SLOS patients.25 26 162
The 2957 bp cDNA for the human DHCR7 gene has an open reading frame of 1425 bp, which codes for a protein with 475 amino acid residues and a calculated mass of 54.5 kDa.24 The gene, which spans 14 kb of genomic DNA, is organised into nine exons and, by hydropathy plot, between six and nine transmembrane alpha helices. In addition to having a high degree of homology with DHCR7 ofArabidopsis thaliana, the humanDHCR7 has substantial homology with genes encoding sterol-delta14-reductases across phyla and with the gene encoding the human lamin B receptor, a nuclear inner membrane protein with intrinsic sterol-delta14-reductase activity but unknown nuclear function or physiological significance.24 146 Among the 26 alleles studied by Fitzky et al,162 only one example of IVS8-1G>C, a splice site mutation that creates a 134 bp insertion between exons 8 and 9, was found. However, study of a cohort of mostly North American patients with largely European and Hispanic ancestry showed that the IVS8-1G>C mutation constituted 29% of DHCR7 mutant alleles, and that several other alleles, R404C (11%), V326L (7%), W151X (8%), and T93M (8%), also occurred at relatively high frequencies.163Collectively, the five most common mutations from this study and the Dutch study group (Waterham et al, unpublished data) accounted for two-thirds of the mutant alleles (fig3). Although these high frequencies may have evolved because of a special heterozygote advantage afforded by the particular mutations, more likely is a combination of founder effect and a heterozygote advantage that encouraged the persistence of these several mutations and of diverse missense mutations. As would be predicted in a model of heterozygote advantage, such as increased synthesis of vitamin D by heterozygotes, the most common mutations cause more severe enzymatic defects than the less common mutations. There is as yet no correlation between individual mutations and their effects on the sterol or vitamin D metabolism in heterozygotes. Such information, however, may provide evidence for increased vitamin D levels as a possible heterozygote advantage, since European palaeolithic studies have indicated that rickets was a common paediatric disease. As anticipated from biochemical studies, the most severely affected patients are either homozygotes or compound heterozygotes for mutations shown or predicted to have no or severely reduced DHCR7 activity.163 In contrast, most classical “type I” SLOS patients are compound heterozygotes for a severe, truncating mutation and a second missense mutation associated with residual enzyme activity. Most patients with very mild clinical and biochemical phenotypes are compound heterozygotes for two unique or uncommon missense mutations.163

{kind=link}
{kind=link}
{kind=link}
Distribution of 122 SLOS mutations of 3beta-hydroxysteroid-delta7-reductase (DHCR7) in 61 patients with Smith-Lemli-Opitz syndrome.
Biochemical diagnosis
DIAGNOSTIC METHODS
Because approximately 10% of patients with SLOS have normal serum cholesterol levels at any age, including at birth, a blood cholesterol level is not a reliable screening test for SLOS. Moreover, because 8DHC and 7DHC react as cholesterol in cholesterol oxidase assay methods, hospital laboratories may report a normal “cholesterol” level even when 7DHC and 8DHC constitute more than half of the sterols measured. In a few affected children, the level of 7DHC may be normal or equivocally increased, but the 8DHC level is usually mildly but distinctly abnormal. In rare cases where the levels of 7DHC and 8DHC are normal or fall in the heterozygote range, eitherDHCR7 mutational testing, DHCR7 enzymatic assay, or analysis of sterol biosynthesis in cultured cells must be undertaken.153 164 165 Under conditions of maximal growth stimulation in cholesterol depleted cultured medium, lymphoblasts from the mild biochemical variants of SLOS still develop markedly increased levels of 7DHC.
Not uncommonly, there is a need to make a retrospective biochemical diagnosis of SLOS in a child for whom there may be remaining stored tissue or laboratory samples. Samples commonly used for retrospective biochemical diagnosis of SLOS include archived plasma and serum samples, Guthrie newborn screening cards, frozen or formalin preserved necropsy tissues, and amniotic fluids. Recovering sufficient sterol for diagnosis from formalin preserved tissue stored more than a few months is not assured, but can still be successful. In a 130 year old case from an Anatomical Museum the lowered cholesterol level could be proven.166 Because 7DHC and 8DHC are oxygen sensitive, diagnostic levels of 7DHC and 8DHC may be lost with storage, but diagnoses using refrigerated Guthrie cards as old as five years have been made.
OTHER CAUSES OF INCREASED LEVELS OF 7DHC
7DHC exists in normal human tissues at a relatively constant ratio to cholesterol. Thus, in common forms of hypercholesterolaemia, such as familial hypercholesterolaemia resulting from abnormalities of LDL metabolism, the absolute level of 7DHC may be increased, but the ratio relative to cholesterol is usually normal. In some other conditions where there is marked upregulation of cholesterol biosynthesis, such as cerebrotendinous xanthomatosis, 7DHC may be increased 10 to 20-fold, but the presence of similarly increased levels of other sterol precursors and cholestanol clearly establish the correct diagnosis. Another cause of mildly increased levels of 7DHC is treatment with haloperidol (Kelley, unpublished observations), which has a high affinity for the DHCR7 substrate binding site.24 Because DHCR7 is a member of the “sigma” class of drug binding proteins,24 it is likely that other drugs of this class will cause increased levels of 7DHC through inhibition of DHCR7. More importantly, however, such drugs may have particularly adverse effects on patients with SLOS, who have already markedly diminished activities of DHCR7.
PRENATAL DIAGNOSIS
Prenatal diagnosis of SLOS has been accomplished in many pregnancies. As reported by several authors,13 95 154one of the early signs of an SLOS fetus is an abnormally low maternal serum level of unconjugated oestriol. Other elements of the triple screen (chorionic gonadotrophin and α-fetoprotein) are also mildly depressed in some SLOS pregnancies. A number of affected fetuses have been identified by the discovery of suggestive fetal abnormalities, such as nuchal oedema, microcephaly, cleft palate, polydactyly, cystic kidneys, ambiguous genitalia, or a 46,XY karyotype in a phenotypically female fetus. Determination of the level of 7DHC in the amniotic fluid is possible, with typically a more than 500-fold increase in affected pregancies.95 In some pregnancies, the amniotic fluid level of cholesterol is abnormally low, whereas in others the level is surprisingly normal. Direct analysis of the sterol composition of chorionic villi at 10 weeks is also a reliable method for diagnosis of SLOS, although the relative increase in the 7DHC/cholesterol ratio is not as great as it is in amniotic fluid.95 The initial experience with more than 100 pregnancies at risk for SLOS has yielded no false negatives and no false positives.95 Despite the feasibility of prenatal diagnosis by molecular testing of villus tissue or amniocytes, the simplicity and accuracy of biochemical testing obviates the need for molecular analysis except, perhaps, in a rare case with equivocal biochemical results.
Management
Until recently, SLOS could only be treated supportively. However, with the recognition and treatment of the deficiency of cholesterol biosynthesis in SLOS, the care of patients has changed substantially. The estimated daily synthetic need for cholesterol during infancy is 30 to 40 mg/kg/day, which decreases to approximately 10 mg/kg/day in adults.167 168 This daily requirement is usually met by a combination of dietary cholesterol and de novo synthesis, balanced in a highly regulated manner. If basic regulation of cholesterol biosynthesis is normal in SLOS children, children with SLOS may be able to absorb sufficient dietary cholesterol to downregulate endogenous sterol synthesis and therefore limit significantly the de novo production of 7DHC. For this reason, children with SLOS routinely have been supplemented with dietary cholesterol with the expectation that blood cholesterol levels will rise to normal at the same time that the abnormal accumulation of 7DHC is eliminated. A complete biochemical “cure” by such therapy would require that dietary cholesterol be distributed to all cellular and tissue compartments. This does not seem to occur, since cholesterol in the central nervous system appears to be synthesised locally and cannot be transported across the blood-brain barrier.133 Even if cholesterol could reach brain cells and myelin, cognitive performance might not improve, since the mental retardation of SLOS appears to reflect more the embryological micrencephaly and other abnormalities of cerebral neuronal development than an existing deficit of cholesterol or toxic effect of 7DHC.
Initial experimental treatment protocols for SLOS provided 50 mg/kg/day cholesterol, and higher doses up to 300 mg/kg/day, either in natural form (eggs, cream, liver, meats, and meat based formulas) or as purified food grade cholesterol, with or without supplementary bile acids.169-171 For substantially growth retarded children, treatment with cholesterol is sometimes followed by striking increases in the rate of growth for several months until more appropriate weight and height centiles are reached. Paradoxically, when SLOS children enter a growth spurt from better nutrition, cholesterol levels typically fall and 7DHC levels rise substantially despite their marked clinical improvement. Although the changes in plasma sterol levels early in a treatment programme usually do not reflect the obvious clinical benefits of cholesterol supplementation, and normalisation of the cholesterol level should not be the ultimate goal in itself, over a period of years all but the most severely affected children usually show a substantial increase in the level of cholesterol and a corresponding fall in the levels of 8DHC and 7DHC. More striking and rapid changes in blood sterol levels were reported in a rat model of SLOS created by treating rats with BM 15 766, a relatively specific inhibitor of DHCR7.172 The study examined the effects of cholic acid and lovastatin, an HMG-CoA reductase inhibitor, on the blood sterol levels and found that cholic acid had little effect. However, when the rats were given lovastatin plus cholesterol, the anticipated fall in 7DHC levels was prevented rather than enhanced. Thus, because of the lack of definitive evidence that 7DHC is “toxic”, and because of the concern that HMG-CoA reductase inhibitors might limit the synthesis of other critical isoprenoid compounds, HMG-CoA reductase inhibitors are not currently used for routine treatment of SLOS. Bile acid supplements have not been included in more recent SLOS treatment protocols. Indeed, because certain bile acids may downregulate tissue levels of LDL receptors, the more rapid rise in plasma cholesterol levels and the persistently high levels of 7DHC in bile acid supplemented patients may reflect impaired tissue uptake of sterols rather than enhanced intestinal absorption of cholesterol.172 When SLOS children are hospitalised for surgery or acute medical problems and cholesterol cannot be given enterally, cholesterol in the form of LDL containing fresh frozen plasma can have striking beneficial effects, especially for treatment of acute infections and poor wound healing.
In addition to the biochemical and physical changes that follow the initiation of cholesterol replacement therapy, there have also been a number of striking behavioural changes. Upon treatment, irritability and sleep disorders may improve in young children with SLOS within days or weeks, whereas tactile hypersensitivities, especially of the hands and feet, take longer to ameliorate. Typically, all of these behaviours return when cholesterol supplementation is stopped. The behavioural improvement does not appear to correlate with any specific change in the plasma sterol profile, and even children with normal or near normal plasma cholesterol levels may show substantial behavioural improvement when given cholesterol supplements. After infancy, many SLOS patients will develop behaviours such as rocking, finger flicking, object twirling, gaze avoidance, and various obsessions, which meet criteria for the diagnosis of autistic disorder and which typically improve or resolve with cholesterol treatment.
A large proportion of patients may require gavage or gastrostomy feeding for many months or years. Fundoplications are frequently unsuccessful in treating gastro-oesophageal reflux. In most instances, the reflux is caused not by intrinsically abnormal gastro-oesophageal function, but by protein allergy or by simple overfeeding from ill advised attempts to make SLOS children with primordial dwarfism grow faster. The combination of gastrointestinal protein allergy, intrinsic intestinal dysmotility, and microgastria often make feeding management extremely difficult; feeding with elemental formulas may be required for many months.
An uncommon but sometimes severe nutritional problem in SLOS is idiopathic hypermetabolism, which most often occurs between the ages of 1 and 3 years. SLOS children who develop this unexplained hypermetabolism may require up to 200 kcal/kg/day to maintain body weight. Because of the complicating factors of microgastria and intestinal dysmotility, such children often do not gain weight for many months, but can otherwise remain healthy. Tests for endocrine abnormalities or intestinal malabsorption have been normal in such cases, but signs of hypermetabolism such as warm skin and tachycardia are often noted.
Although many SLOS patients have a severe deficiency of normal bile acids, fat malabsorption and deficiencies of fat soluble vitamins are not common in SLOS, and also clinical evidence of steroid hormone deficiency is surprisingly uncommon.96 Among these children, the most common finding is a mild to moderate deficiency of aldosterone synthesis, usually manifest as hyponatraemia and hyperkalaemia during the added hormonal stress of an infection. Glucocorticoid response to infection or to administered ACTH can also be subnormal.96 However, for most SLOS children glucocorticoid production appears to be normal and supplements during infections or perioperatively are probably not needed. Nevertheless, the potential for developing mineralocorticoid and glucocorticoid deficiency postoperatively or during an acute illness must be remembered.
In conclusion, SLOS may now be considered the example par excellence of metabolic dysmorphology. It is well defined biochemically, enzymatically, and molecularly. However, many parts of the pathogenesis, that is, the developmental pathways involved, still remain to be elucidated, and much work has to be done to reach optimal therapeutic regimens. SLOS was the first malformation syndrome in which a cholesterol metabolism disturbance was found. There are now a number of other entities with such a defect, including Conradi-Hunermann syndrome, CHILD syndrome, desmosterolosis, Greenberg dysplasia, and mevalonic aciduria. Together, these entities constitute a group of metabolic disorders which might be called cholesterolopathies.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵