Article Text

Abstract
Achondrogenesis II-hypochondrogenesis and severe spondyloepiphyseal dysplasia congenita (SEDC) are lethal forms of dwarfism caused by dominant mutations in the type II collagen gene (COL2A1). To identify the underlying defect in seven cases with this group of conditions, we used the combined strategy of cartilage protein analysis andCOL2A1 mutation analysis. Overmodified type II collagen and the presence of type I collagen was found in the cartilage matrix of all seven cases. Five patients were heterozygous for a nucleotide change that predicted a glycine substitution in the triple helical domain (G313S, G517V, G571A, G910C, G943S). In all five cases, analysis of cartilage type II collagen suggested incorporation of the abnormal α1(II) chain in the extracellular collagen trimers. The G943S mutation has been reported previously in another unrelated patient with a strikingly similar phenotype, illustrating the possible specific effect of the mutation. The radiographically less severely affected patient was heterozygous for a 4 bp deletion in the splice donor site of intron 35, likely to result in aberrant splicing. One case was shown to be heterozygous for a single nucleotide change predicted to result in a T1191N substitution in the carboxy-propeptide of the proα1(II) collagen chain. Study of the clinical, radiographic, and morphological features of the seven cases supports evidence for a phenotypic continuum between achondrogenesis II-hypochondrogenesis and lethal SEDC and suggests a relationship between the amount of type I collagen in the cartilage and the severity of the phenotype.
- type II collagen disorders
- achondrogenesis II-hypochondrogenesis
- spondyloepiphyseal dysplasia congenita
- COL2A1
Statistics from Altmetric.com
- type II collagen disorders
- achondrogenesis II-hypochondrogenesis
- spondyloepiphyseal dysplasia congenita
- COL2A1
Several heritable osteochondrodysplasias have now been recognised as members of the family of type II collagen disorders, all of which result from dominant mutations in theCOL2A1 gene.1-4 Phenotypes within this group range from severe lethal dwarfism at birth to relatively mild conditions with precocious osteoarthrosis and little or no skeletal growth abnormality.2 Achondrogenesis II-hypochondrogenesis and lethal spondyloepiphyseal dysplasia congenita (SEDC) represent the more severe end of the spectrum.1 5-7 These entities are characterised by severe disproportionate short stature of prenatal onset. The distinction between these phenotypes is mainly based upon clinical, radiographic, and morphological features but considerable phenotypic overlap often hampers proper classification.1 6-8
All of the previously reported cases of achondrogenesis II-hypochondrogenesis in which the molecular defect was found were heterozygous for a mutation in the COL2A1gene resulting in a glycine substitution in the triple-helical domain of the proα1(II) collagen chain.9-16 Biochemical analysis of cartilage usually showed the presence of type I collagen and post-translationally overmodified type II collagen. Studies on cultured chondrocytes from one case of hypochondrogenesis with a G913C mutation showed lack of secretion of type II collagen in the medium.16 Mutations reported in cases of SEDC are more heterogeneous. Single amino acid substitutions, deletions, or duplications in the triple helical domain of the proα1(II) collagen chain have been reported.17-24
The aim of this study was to identify the mutation in theCOL2A1 gene in seven cases of achondrogenesis II-hypochondrogenesis or severe SEDC based on the results of the biochemical analysis of cartilage tissue. Instead of performing a “head to tail” mutation analysis of the entire gene, we first started screening those regions of theCOL2A1 gene in which a mutation was most likely to reside, based on the electrophoretic properties of the extracted cartilage collagen. The phenotype of each case was studied in relation to the molecular and biochemical findings and compared with other reported cases of achondrogenesis II-hypochondrogenesis and SEDC in which aCOL2A1 mutation was identified.
Materials and methods
PATIENTS
The seven patients in this study were referred in the period 1982 to 1991 to the International Skeletal Dysplasia Registry at Cedars-Sinai Medical Center, Los Angeles for diagnosis. The clinical features and radiographs were sent to the Registry for evaluation and tissue specimens, taken during necropsy, were collected for morphological, biochemical, and molecular studies. All patients were sporadic. Parental DNA samples were not available.
PROTEIN ANALYSIS
Cartilage stored at −70°C was used. For analysis of pepsin solubilised collagen, cartilage was extracted, washed, and digested with pepsin as described previously.11 The resulting collagen type II molecules were analysed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Human epiphyseal cartilage collagen from a 4 day old neonate without signs of a skeletal dysplasia was used as a control. For peptide analysis, cartilage was digested with cyanogen bromide (CB) and the resulting collagen CB peptides were analysed by SDS-PAGE and stained with Coomassie blue as described previously.11
NUCLEIC ACID ANALYSIS
Genomic DNA was isolated from cultured skin fibroblasts by standard procedures. PCR amplifications were performed in 25 μl reactions containing 100 ng of genomic DNA, 2 UTaq DNA polymerase, 2.5 pmol of each primer, 200 μmol/l of each dNTP, 2 mmol/l MgCl2, 10 mmol/l Tris-HCl, pH 8.3, and 50 mmol/l KCl. The amplification conditions consisted of an initial two minutes at 95°C followed by 36 cycles of 95°C for one minute, 60°C for one minute, and 72°C for one minute, followed by a nine minute incubation at 72°C. Primer sequences were 5′-CTGTTCTCACTCACT GCCTC-3′ and 5′-GGATACCATGTGACC TCAG-3′ for exon 22; 5′-GGTTGATCACTT CTTGGTG-3′ and 5′-CTCAGTGGGACTC CAGGCTAC-3′ for exon 31; 5′-GTGCCC GGCTGAGGCGGCTG-3′ and 5′-TCCTAA TGCCCAGCAGTCCAG-3′ for exon 33; 5′-ACCTTCTTACCCCAGCTCTTC-3′ and 5′-GGCCTCGGGCAGAGCCAGGC-3′ for exon 35; 5′-CCCTGACCTGACTCAATC GG-3′ and 5′-AGGAGGCCTCGGGAAGT CCC-3′ for exon 46; and 5′-TCCTCTGA GCTTGCTCCACTC-3′ and 5′-TCCTGTC ACTTTAGGACCTG-3′ for exon 51. Primer sequences for the other exons are available on request. The PCR generated fragments were examined on non-denaturing 6% polyacrylamide gels (29:1 acrylamide:bis ratio) in 1 × TBE buffer. For single strand conformation polymorphism (SSCP) analysis, 5 μCi of α-[32P] dCTP was added to the PCR reaction and the amount of unlabelled dCTP was reduced to 2.5 μmol/l. Fragments were separated by electrophoresis through MDE gels (AT Biochem) for 11 hours at 3.5 W. The gels were dried and exposed tox ray film for 48 hours at −70°C with an intensifying screen. For heteroduplex analysis, PCR products were denatured at 98°C for five minutes, renatured for 60 minutes at 68°C, and loading buffer (50% sucrose, 0.1% bromophenol blue, 0.1% xylene cyanol in water) was added. The samples were separated on MDE gels (1 × MDE gel supplemented with 10% glycerol in 0.6 × TBE) overnight at 250 V. The fragments were visualised by ethidium bromide staining. For DNA sequence analysis PCR generated fragments were purified using a Qiaquick spin PCR purification kit (Qiagen). The purified fragments were either used for direct sequence analysis or ligated into the TA cloning kit (Invitrogen) and sequenced using Sequenase 2.0 (USB). For direct sequencing, single stranded DNA fragments were generated in a second round of amplification using a purified aliquot (2 μl) from the first PCR and only one of the two primers (50 pmol).
CARTILAGE MORPHOLOGY
For light microscopy (LM), cartilage specimens were fixed in 10% formalin and embedded in methyl methacrylate. Five μm sections were cut and stained as described previously.15 For electron microscopy (EM), cartilage specimens were fixed in 2.5% glutaraldehyde, post-fixed in osmium, and dehydrated and embedded in Spurr resin. Thin sections were cut and stained as described previously.15
Results
The diagnosis of a lethal type II collagen disorder was made in each of the patients based on the clinical, radiographic, and morphological features (table 1, fig 1).1 Significant findings on which the diagnosis was based included the following. (1) Clinical: moderate to severe short limbed dwarfism at birth with respiratory problems owing to a small thorax; cleft palate. (2) Radiographic: incomplete ossification of the vertebral bodies; severe shortening of the long bones and ribs; flared iliac wings; absent ossification of pubic bones, talus, and calcaneus. (3) Morphological: hypervascular resting cartilage; chondrocytes with inclusion bodies (LM) or dilated cisternae of the rough endoplasmic reticulum (RER) containing granular material (EM); disorganised growth plate characterised by extension of hypertrophic cells into the primary trabeculae.
Clinical, radiographic, and morphological features of seven patients with a lethal type II collagen disorder

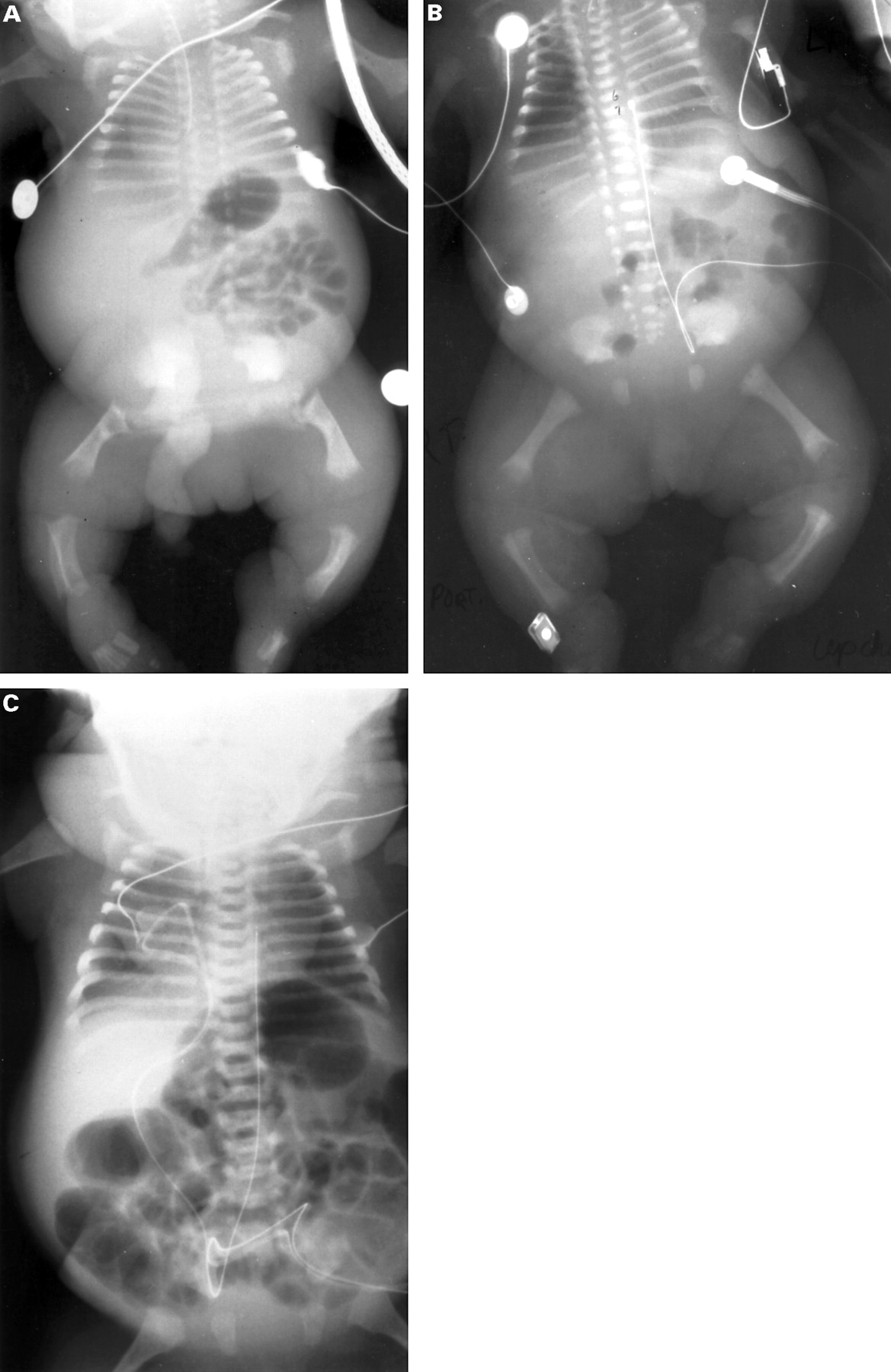
Anteroposterior radiographs of patients R86-153, R83-32, and R91-68. (A) Patient R86-153 represents a severe phenotype characterised by short tubular bones with metaphyseal irregularities, minimal ossification of vertebral bodies in the thoracic spine, hypoplastic iliac wings with flat acetabular roofs, and short ribs. (B) Patient R83-32 shows an intermediate phenotype. (C) Patient R91-68 illustrates a milder phenotype with normal metaphyses of shortened tubular bones, flattened but normally ossified vertebral bodies, normal acetabular roofs, and a less narrow chest.
SDS-PAGE of cyanogen bromide (CB) digested cartilage from patient tissues showed slowly migrating, post-translationally overmodified type II collagen CB peptides not seen in the control (fig 2A). The cases varied in their degree of overmodification as best shown by the extent to which α1(II) CB10 was retarded. CB10 was most retarded for R85-109 and R86-153 and less so for R86-6. From R86-6, however, peptide α1(II) CB11 was noticeably retarded compared with control CB11. A doublet for CB10 was visible in R82-121 and an abnormally broad band for CB10 was apparent in R91-68. These observations suggested where along the proα1(II) collagen molecule the mutation may lie, since post-translational overmodification of the procollagen molecules starts at or near the mutation site and proceeds in an amino-terminal direction. For R86-6, a defect in the middle of the triple helix, amino-terminal to CB10 was postulated, whereas for the other patients a defect within or carboxyl-terminal to CB10 was most likely (fig 2C). SDS-PAGE of pepsin extracted collagen from the patient cartilage also showed slowly migrating, overmodified α1(II) chains in most cases compared with the control (fig 2B). The pepsin extract of R85-109 showed a retarded β component not present in the control extract. The band disappeared on reduction with dithiothreitol (not shown) indicating that it was a disulphide bonded β dimer. Type I collagen was also present in many samples as seen from the CB peptide patterns and from the characteristic pattern of three β dimer bands evident in the pepsin digests. However, type I collagen can be present in dissected samples of normal human neonatal cartilage and the amount can vary between adjacent samples from the same person. Based on the intensity of the α2(I) CB3-5 peptides, type I collagen seemed to be most abundant in patients R83-32, R86-153, and R87-75 (fig 2A).

(A) SDS-polyacrylamide gel electrophoresis of cyanogen bromide (CB) peptides extracted from cartilage of each patient and a control (4 day old neonate).The position of the CB peptides derived from control tissue is shown on the left side for the α1(II) collagen chains and on the right side for the α1(I) and α2(I) collagen chains. The major α1(II) CB peptides (CB10 and CB11) derived from patient's tissue exhibit delayed mobility compared to the control. Based on the intensity of the α2(I) CB3-5 peptides, type I collagen seems to be most abundant in patients R83-32, R86-153, and R87-75. (B) SDS-polyacrylamide gel electrophoresis of pepsin extracted collagen from cartilage of each patient and a 4 day old neonate (control) shows slowly migrating, overmodified α1(II) chains in most patients compared with the control. The pepsin extract of R85-109 shows a retarded β component (arrow) not present in control extract. The position of normal α1(II) chains, α1(I) chains, α2(I) chains, and β dimers is indicated on the right side. (C) Cyanogen bromide peptide map of proα1(II) collagen. The corresponding exons and positions of methionine residues are shown at the top.
Since in the majority of the cases a defect was postulated to reside within or carboxyl-terminal to α1(II) CB10, the region corresponding to exons 32-49 of theCOL2A1 gene was first targeted for mutation analysis (fig 2C). PCR generated genomic DNA fragments for exons 32-49 were examined by SSCP analysis. Primers were designed to include the intron-exon boundaries in each exon amplification. SSCP analysis showed an abnormal pattern for fragments containing exon 46 in patients R85-109 and R86-153 (fig 3), and for fragments containing exon 35 in patient R91-68 (fig 4). Direct sequence analysis of the PCR products of exon 46 showed the mutation in each of the two patients (fig 3). Patient R85-109 was heterozygous for a G to T transversion at the first nucleotide of exon 46. This change implied substitution of glycine910 by cysteine and abolished aBstNI restriction endonuclease site. Patient R86-153 was heterozygous for a G to A transition, predicted to result in substitution of glycine943 by serine. The mutation abolished a cleavage site for the restriction endonucleaseMspI. Cleavage of the amplified DNA fragments with the corresponding restriction endonucleases was used to confirm that both patients were heterozygous for the mutation (data not shown). DNA sequence analysis of clones containing exon 35 showed a 4 bp deletion at the donor splice site of intron 35 in patient R91-68 (fig 4). Neither cultured cells nor frozen cartilage with which to test the prediction of a splicing mutation were available.

Identification of the COL2A1 mutation in patients R85-109 and R86-153. (Top) SSCP analysis of amplified genomic DNA fragments containing exon 46. The arrows indicate the abnormally migrating fragments from R86-153 (lane 1) and R85-109 (lane 5) compared to unrelated controls (lanes 2-4, 6). (Bottom) Partial nucleotide sequence of exon 46 (upper case) and intron boundaries (lower case) containing both point mutations. For R85-109, the G to T transversion predicts substitution of cysteine for glycine at residue 910. For R86-153, the G to A transition implies substitution of serine for glycine at residue 943. The restriction endonuclease cleavage sites disrupted by the mutation are underlined.

Identification of the COL2A1 mutation in patient R91-68. (Top) SSCP analysis of amplified genomic DNA fragments containing exon 35. The arrow indicates the abnormally migrating fragment from patient R91-68 (lane 5) compared to unrelated controls (lanes 1-4, 6-8). (Middle) Identification of the 4 bp deletion at the splice donor of intron 35. The sequence was determined after cloning of PCR products, containing exon 35 (upper case) and intron (lower case) boundaries, from the mutant and normal alleles. Five of eight clones showed the 4 bp deletion at the splice donor of intron 35. (Bottom) The IVS35+4delAGTG mutation implies skipping of exon 35.
For identification of the underlying defect in the four other patients, the same region (exons 32-49) was first re-evaluated by heteroduplex analysis and thereafter this analysis was extended to the other part of the triple helical domain of the proα1(II) chain, corresponding to exons 6-31. Only for patient R83-32 did polyacrylamide gel electrophoresis of PCR generate products containing exon 33 showing both normal and more slowly migrating fragments in a position which suggested interallelic heteroduplexes. DNA sequence analysis of clones containing exon 33 showed a G to C transversion, implying substitution of glycine571 by alanine (fig 5). The mutation did not affect a cleavage site of any known restriction endonuclease. Restudy of the earlier performed SSCP analysis of amplified DNA fragments containing exon 33 in patient R83-32 did not show any abnormalities. Taking into account the known lack of 100% sensitivity of SSCP and heteroduplex analysis in mutation detection, direct sequence analysis of PCR amplified genomic DNA fragments spanning the entire triple helical domain was performed in the remaining three patients. This study showed that patient R86-006 was heterozygous for a G to A transversion, predicted to result in substitution of glycine313 by serine and that patient R87-75 was heterozygous for a G to C transition, predicted to result in substitution of glycine517 by valine. These mutations did not create or destroy a restriction endonuclease cleavage site. No sequence changes in the triple helical domain were found in patient R82-121. However, in this patient, a C to A transversion at the second nucleotide of codon 1191 was identified. This single nucleotide change implies substitution of threonine1191 by asparagine (T1191N) in the carboxy-propeptide of the proα1(II) collagen chain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Identification of the guanine to cytosine transversion in exon 33 of the COL2A1 gene for patient R83-32. Partial nucleotide sequence of a clone containing the normal and mutant allele is shown. Three of 10 clones showed the nucleotide change.
Discussion
Identification of the molecular defect in patients with type II collagen disorders is usually a challenge because of the relatively large size and complexity of the COL2A1 gene and its main expression in cartilage. Cartilage is not easily accessible, relatively acellular, and therefore a poor source of mRNA for cDNA synthesis and analysis. Furthermore, sufficient amounts of mRNA can often not be obtained from cultured skin fibroblasts or lymphocytes owing to low basal transcription of the gene. Therefore, mutation analysis of the COL2A1 gene is often performed in the genomic DNA in a “head to tail” fashion, which is time consuming and laborious. In this study, we used the opportunity of having cartilage tissue for exploring the genetic defect in the COL2A1 gene in seven patients with a lethal type II collagen disorder. Those regions of theCOL2A1 gene in which a mutation was likely to reside, based on the overmodification pattern of the type II collagen CB peptides, were screened first with SSCP and heteroduplex analysis. Using these mutation detection methods, the underlying defect was found in four of seven patients. The mutation in two other patients (R86-6 and R87-75) could only be identified with direct sequence analysis of PCR amplified genomic DNA fragments.
The clinical, radiographic, and morphological features of each patient were consistent with the diagnosis of a lethal type II collagen disorder (table 1, fig 1). Significant overlap in phenotype was noticed (table 1). With the exception of patient R91-68, who represents severe SEDC rather than achondrogenesis II-hypochondrogenesis because of the complete ossification of the spine and less severe shortening of the long bones, all cases fell into the achondrogenesis II-hypochondrogenesis spectrum. In comparison to the other cases, the phenotype of patient R82-121 was remarkable, radiographically by more metaphyseal involvement and clinically by absence of respiratory insufficiency in the postnatal period. The baby died at 13 months following seven months' hospitalisation for severe, bilateral bronchopneumonia.
The results of the biochemical analysis showed post-translationally overmodified type II collagen in significant amounts in each patient's cartilage. This is consistent with previous findings that mutant α1(II) chains can be secreted and incorporated into the cartilage matrix in patients with achondrogenesis II-hypochondrogenesis or SEDC.11 15 18 24 However, in one case of hypochondrogenesis with a G913C mutation, cultured chondrocytes failed to secrete α1(II) chains in the medium.16 Type I collagen was also present in the cartilage tissue of each patient and appeared to be increased in R83-32, R86-153, and R87-53, who are the most severely affected (table 1). This might suggest a relationship between the amount of type I collagen in the cartilage and the severity of the phenotype. Probably in response to insufficient amounts of type II collagen in the extracellular matrix, the chondrocytes produce and secrete more type I collagen as a substitute. One study has shown that the genes for type I collagen are expressed in chondrocytes of a patient with hypochondrogenesis.12 This would suggest that hypervascularity of the cartilage is not the main explanation for the presence of type I collagen in cartilage of these patients.
It has been postulated that mutations in the triple helical domain of procollagen α chains delay the folding of the protein into the triple helical conformation, which causes post-translational overmodification of the protein amino-terminal to the mutation site.25 The availability of cartilage from each case allowed us to screen for an overmodification pattern of the CB peptides and so direct the molecular analysis of the COL2A1 gene based on the biochemical findings. Because of the slower mobility of peptide CB10 in all but one of the seven cases, it was decided to explore first the domain of COL2A1 encoding exons 32-49 (fig2C).
Analysing these exons using SSCP and heteroduplex analysis, three patients (R86-153, R85-109, and R83-32) were found to be heterozygous for a missense mutation that predicted substitution of glycine in the triple helical domain of the α1(II) chain. Their phenotype resided at the more severe end of the achondrogenesis II-hypochondrogenesis and SEDC spectrum. In patient R85-109, with a G910C mutation, the mutant α1(II) chains were clearly expressed in the tissue as seen from the presence of a reducible β dimer in the pepsin extracted collagen (fig2B). The peptide patterns for R85-109 and R86-153 were remarkably similar for both pepsin and CB digests of tissue and it is notable that the causative single amino acid substitutions (G910C and G943S) were located near the carboxyl-terminus of the triple helix. Interestingly, the G943S mutation, identified in patient R86-153, has been previously identified in another, unrelated case.9 25 26 The recurrence of the mutation could be explained by its occurrence in the context of a CpG dinucleotide. The clinical, radiographic, and morphological features of both cases were almost identical illustrating the reproducible pathogenetic effects of this mutation (table2).9 25 26
Clinical, radiographic, and morphological features of two patients with the G943S mutation in the COL2A1 gene
Analysis of the remaining exons 6-31 of the triple helical domain of the proα1(II) collagen chain by heteroduplex analysis did not identify any abnormalities in patients R82-121, R86-6, and R87-75. However, direct sequence analysis of PCR amplified genomic DNA fragments from this region showed a G313S mutation in R86-6 and a G517V mutation in R87-75. As predicted by the overmodification pattern of the CB peptides, the mutation in R86-6 did indeed reside amino-terminal to CB10. Comparable to patients R85-109 and R86-153, the peptide patterns for patients R83-32 and R87-75 were also quite similar. Again, the mutations (G517V and G571A) were located in the same region around the amino-terminal border of CB10. Direct sequence analysis of PCR generated genomic DNA fragments for exons 6-49, spanning the entire triple helical domain, did not show any abnormalities in patient R82-121, despite the observed doublet for CB10. The significance of the single nucleotide change, located in the carboxy-propeptide and predicted to result in substitution of threonine1191 by asparagine (T1191N), remains unclear. Mutations in the carboxy-propeptide of the proα1(II) collagen chain have not been reported before in patients with a lethal type II collagen disorder. However, single amino acid substitutions in the carboxy-propeptide of the proα1(I) collagen chain have been described in osteogenesis imperfecta type II, indicating that mutations in this region can have a dramatic effect on the function of a fibrillar collagen molecule.27
In patient R91-68, a 4 bp deletion was found at the donor splice site of intron 35. Although the possibility of exon skipping could not be investigated at the cDNA level, the overmodification of the type II collagen CB peptides (fig 2A) indicates that an abnormal α1(II) chain was expressed and incorporated into the matrix. We concluded that the mutation probably resulted in skipping of exon 35 with shortened chains being incorporated into the tissue fabric. Consistent with the published findings of others, the phenotype of this case was considerably milder than that of the three cases with a glycine substitution.2 It is notable that most cases of Kniest dysplasia, which is usually a non-lethal type II collagen disorder, are caused by COL2A1 exon skipping mutations.28-31 These deletions tend to be concentrated in the amino-terminal half of the molecule. Since the type II procollagen molecule consists of three identical α1(II) chains, heterozygosity for a missense mutation in theCOL2A1 gene should result in seven/eight procollagen trimers containing one, two, or three mutated α1(II) chains. A mutation in one COL2A1 allele affecting mRNA splicing and resulting in an in frame deletion of several amino acids will give rise to six/eight procollagen trimers containing a mixture of normal and deletion containing chains. Two/eight of the molecules will contain either three normal or three shortened α1(II) chains. This could explain to some extent the difference in phenotypic outcome between glycine substitutions and in frame deletions as a result of aberrant splicing. However, not all glycine substitutions in the α1(II) chain result in a severe phenotype as they can also cause milder cases of SEDC and phenotypically milder spondyloepimetaphyseal dysplasia Strudwick type.2 32 The phenotype probably depends not only on the mutation type (for example, exon deletion or glycine substitution), but also on the local domain of the triple helix affected.
Acknowledgments
We thank clinical colleagues M Bridge, N Chaney, A Donnenfield, D Huff, M Lipson, H Stern, and J Waterson for referring the patients described in this study. Excellent organisational assistance was provided by M Priore and S Levin of the International Skeletal Dysplasia Registry. This work was funded by NIH grants AR37318 (to DRE) and HD22657 (to DLR, DHC, and RSL) and by a Belgian National Fund for Scientific Research grant G.0013.97 (to GRM, LN, and ADP). The authors declare that the experiments comply with the current laws of the country in which the experiments were performed and that the study was approved by the Ethical Committee.
References
Linked Articles
- Correction