Article Text

Abstract
A large number of cases with supernumerary marker chromosomes (SMCs) should be compared to achieve a better delineation of karyotype-phenotype correlations. Here we present four phenotypically abnormal patients with autosomal marker chromosomes analysed by fluorescence in situ hybridisation using centromeric, telomeric, and unique sequence probes, as well as forward and reverse painting. We also report the first case, to the best of our knowledge, of an SMC derived from chromosome 5. Furthermore, a marker chromosome 20 in a patient with sex differentiation abnormalities, a double mar(6) in a boy with psychomotor retardation, and the association of r(19) with dup(21q21.2q22.12) are described. Although the mar(6) was very small, the presence of euchromatin was shown, suggesting that the partial trisomy of pericentric region derived sequences is implicated in the aetiology of the abnormal phenotypes.
- supernumerary marker chromosomes
- fluorescence in situ hybridisation
- phenotype-genotype correlation
Statistics from Altmetric.com
Until recently, supernumerary marker chromosomes (SMCs) have presented a difficult diagnostic problem for cytogeneticists and for clinicians. Introduction of fluorescence in situ hybridisation (FISH) into chromosome analysis has allowed chromosome origin, structure, and in some cases genetic content of marker chromosomes to be identified. Many SMCs have already been characterised using FISH.1-9Nevertheless, because of the great variety and a scarcity of SMCs derived from some of the autosomes, many of them still need to be characterised further. Except for inv dup(15) and inv dup(22), for which phenotypic consequences are now well known, most SMCs require more cytogenetic and clinical data for further insight into karyotype-phenotype correlations.10-17 A recent publication by Crolla8 showed the importance of accumulating a large amount of FISH data for various marker chromosomes in relation to their phenotypic effects for reliable risk estimation. The author calculated from the review of data on non-acrocentric autosomal SMCs that the risk of an abnormal phenotype for this group is ∼28%. This is twice as high as the risk previously estimated based on the results of a large sample of prenatal studies using conventional methods.18
Up to now, supernumerary markers derived from all the autosomes except for chromosome 5 have been identified. In rare cases, more than one SMC was found in the same person.19-23 We present four cases with non-acrocentric supernumerary autosomal markers including the first case of SMC(5) and a case with two SMCs(6).
Materials and methods
All four cases with supernumerary marker chromosomes were referred to our laboratory specifically for FISH studies of markers found in other cytogenetic units. In all cases, the karyotype using conventional cytogenetic analysis following GTG banding was reassessed.
FISH was performed on conventional, 1 day to 6 month old chromosome preparations. All markers were analysed using digoxigenin labelled alphoid centromere specific and all telomeric probes from Oncor (Gaithersburg). Biotinylated chromosome specific libraries from Cambio (Cambridge) or Chromoprobe-M Multiprobe directly labelled with cyanine 3 (Cy3) from Cytocell were also applied in all cases. The unique sequence probes were chosen using Genome Database GDBclone query(http://www.hgmp.mrc.ac.uk/gdb/gdbtop.html) and Resources for Human Molecular Cytogenetics (http://bioserver. uniba.it/fish/Cytogenetics/welcome.html).
Isolated probe DNA and Alu-PCR products of YAC 882A10 were prepared and labelled by nick translation reaction using biotin labelled nucleotides (Life Technologies, Gibco, BRL). A standard in situ hybridisation protocol according to the methods of Pinkel et al 24 and the recommendations of the manufacturers of the probes was used. Digoxigenin was detected using fluorescein isothiocyanate (FITC) conjugated layers of antibodies (Oncor) and biotin was visualised with two layers of FITC-avidin DCS (Vector Labs). Chromosomes were counterstained with DAPI and propidium iodide (PI) diluted in Vectashield antifade (Vector). Metaphases were analysed under a Nikon epifluorescence microscope equipped with appropriate filter combinations. For documentation, photomicrographs were obtained with Kodak 400 or 1000 ASA negative using dual exposure. Digital image system including Axioplan epifluorescent microscope (Carl Zeiss) equipped with cooled CCD KAF 1400 S200 camera and IPLab Spectrum software (Photometrics) were also used for documentation of the results of FISH studies.
Chromosome microdissection, DOP-PCR, and subsequent reverse painting were performed as previously described.25
Microsatellites for UPD studies had been selected using GDBamplimer query and were amplified using routine methods with PCR primers obtained from InViTek (Berlin). The products were separated on 8% polyacrylamide gels and visualised by the silver staining method.
The loci location, order, and SMC sizes were inferred from The Genetic Location Database LDB (http://cedar.genetics.soton.ac.uk/public_html/) and GDB maps.
Cell lines from the probands are not available.
PATIENTS
Case 1 (SMC 5)
This 27 year old women was referred to the Internal Medicine Department for painful oedema of the lower leg. Because of her small facial features, epicanthic folds, and high arched palate, karyotype analysis was performed. She was born after an uncomplicated pregnancy. Her mental development was slightly delayed and she began to speak indistinctly at the age of 4 years. Physical examination showed hypertelorism, left strabismus, upward slanting palpebral fissures, synophrys, prominent nose with wide nasal bridge, micrognathia, low set, posteriorly rotated, dysplastic ears with abnormal helix and lobule, and low frontal and posterior hairline (fig 1). Furthermore, bilateral talipes valgus and genu valgum with restriction of active and passive movement of the knee joints were found.X ray, arthrography, and ultrasonography studies showed bilateral narrowing of their articular spaces, the presence of cysts linked with the joint cavities, and pathological serofibrinous exudate. Bilateral ruptured Baker's cysts of the knees resulting in the posterior compartment syndrome were diagnosed. They probably arose from the inborn flexibility of the articular capsules since rheumatoid arthritis as well as other causes of Baker's cysts were excluded. Synovectomy was performed with a good result. On follow up at the age of 30 years, she was found to be mildly mentally retarded.

Case 1 aged 30 years. Note epicanthic folds, hypertelorism, left strabismus, upward slanting palpebral fissures, and prominent nose with a wide nasal bridge.
Case 2 (SMC 6)
A 2 year old boy was referred because of psychomotor retardation, hypotonia, congenital heart defect (VSD and PDA), and mildly dysmorphic facial features.
Case 3 (SMC 19)
The marker chromosome was found during amniocentesis for advanced (43 years) maternal age. The male proband was born by caesarian section at 40 weeks' gestation following an uncomplicated pregnancy. At birth, physical examination showed hypotonia, dysmorphic craniofacial features including microcephaly, hypertelorism, and upward slanting palpebral fissures, and deep palmar and plantar creases. MRI showed delayed myelinisation and agenesis of a splenium of the corpus callosum. His psychomotor development at 9 months of age was moderately delayed.
Case 4 (SMC 20)
The female proband was referred to the Endocrinology Department at the age of 18 years because of primary amenorrhoea, hirsutism, and mild mental retardation. On physical examination, myopic retinal degeneration and horizontal nystagmus were found. Ultrasonography showed absence of the uterus, a 35 mm cyst with irregular reflections in the left gonad, and abnormal ostium of the ureters in the bladder. The second gonad was not found.
Results
CYTOGENETIC, FISH, AND MICROSATELLITE STUDIES
The major clinical findings and cytogenetic data of the marker chromosomes are shown in table 1. The results of the FISH and microsatellite polymorphisms are summarised in table 2.
The major clinical traits and cytogenetic characteristics of marker chromosomes
Summary of the results of the FISH and microsatellite polymorphism analysis in cases 1-4 (A–D, respectively)
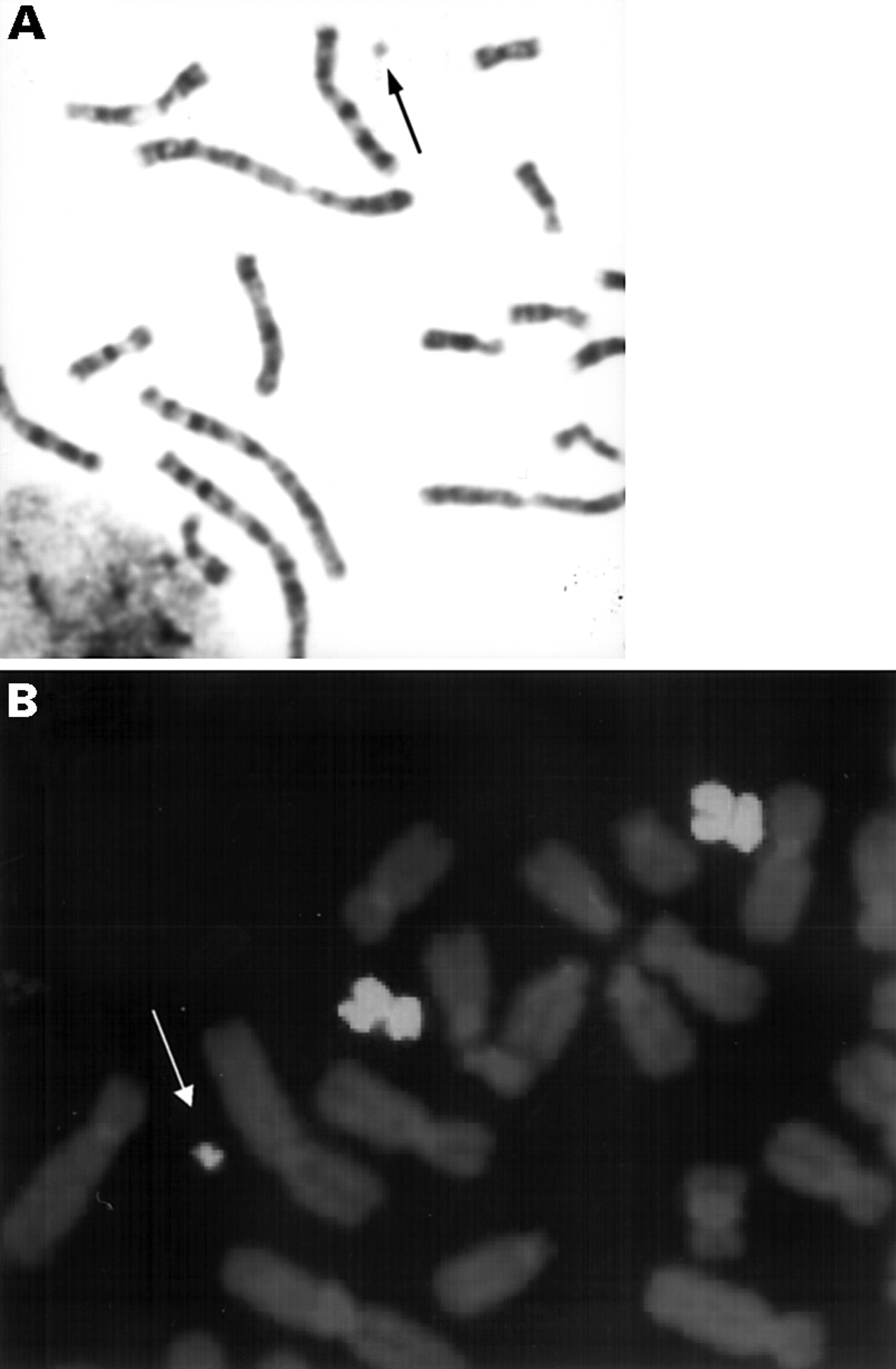
Case 1: 47,XX,+r[39]/46,XX[29].ish r(5)(?p14?q11.1)(wcp5+,D5S2520+, D5S430+,D5S427/D5S668−,D5S684−,D1Z7/D5Z2/D19Z3+).rev ish r(5)(p14q11.2)
A small, ring shaped, supernumerary marker chromosome was found in 57% of metaphases analysed. The parental origin of the marker could not be established. Reverse painting indicated that the genetic content of the ring extended from band p14 to q11.2 of chromosome 5 (fig 2). No double ring forms were observed using D1Z7/D5Z2/D19Z3, D5S2520, and D5S430 specific probes. Microsatellite polymorphism analysis was informative for two distal loci, D5S208 and D5S352, and allowed exclusion of uniparental disomy of chromosome 5. No alleles from the second maternal homologue 5 were found in the 5p14-q11.2 region and the paternal bands of PCR products at D5S418 and D5S398 were fainter than the maternal ones, indicating that r(5) arose from maternal chromosome 5.

(A) G banded partial karyotype and (B) the result of microdissection of r(5) and subsequent reverse painting in case 1. Arrowheads indicate 5p14-q11.2 origin of r(5).
Case 2: 48,XY,+2mar[59]/47,XY,+mar[25]/46, XY[40] de novo.ish 2der(6)(p11.1q12)(wcp6+, D6S159−, D6S173−,D6S182−,GSTAP1−, D6S466/ D6S1623/D6S1573−,D6S147+, D6S150−,D6S151−,D6Z1+)
One or two minute additional marker chromosomes were found in 68% of peripheral blood lymphocytes. Two markers were observed in 47% of metaphases. The parental karyotypes were normal. In both cell lines, the markers were positive with the centromeric probe specific for chromosome 6. The chromosome 6 paint and D6S147 (6q12) unique sequence probe also gave positive results indicating the presence of euchromatin (fig 3). The same morphology and FISH characteristics of the markers suggests a duplication event resulting in the cell line with two markers. Since the proband's father was not available for DNA studies his parents were analysed. Uniparental disomy of chromosome 6 was excluded using microsatellite polymorphism analysis at D6S311 andER-1/ER-2.

(A) G banded chromosomes and (B) FISH with cosmid cCI6-21 (D6S147) in case 2. SMCs(6) are indicated with arrows.
Case 3: 46,XY,dup(21)(pter→q21.2::?q22.2→ q21.2::q21.2→qter)[32]/47,XY,dup(21) (?q22.2q21.2),+ mar[18]de novo.ish r(19)(p13.11q13.2)(wcp19+,D19S11−, D19S452+,D19S269+, D19S752+,D19S190+, D1Z7/D5Z2/D19Z3+),dup(21)(?q22.12q21.2) (wcp21+,APP++,D21S58+,contig21q22.3+)
A small Ag-NOR and distamycin A/DAPI negative marker chromosome was found in 18% of amniocytes. Parental chromosomes were normal. Postnatal verification of the karyotype in peripheral blood lymphocytes carried out in the same cytogenetic laboratory confirmed the presence of the marker chromosome. Verification of the karyotype with banding techniques in our laboratory showed additionally an abnormal chromosome 21 with inverted duplication of q21.2→?q22.2 in all cells examined. Results of cytogenetic-molecular analysis of abnormal chromosomes are shown in fig 4. Insertion from the other chromosome was excluded using a chromosome 21 painting probe (Cambio) and dup(21q) confirmed with probe APP (22q22.1). FISH with “Down syndrome cosmid contig” (21q22.3) (Cambio) and D21S58 (22q22.12) specific probes excluded the duplication of the Down syndrome critical region (DSCR) in 22q22.2.

(A) Abnormal chromosomes from case 3 identified as r(19) and dup(21) after G banding and (B) after hybridisation with the whole chromosome paint 19. (C) Cosmid probes 15694 (D19S752) and (D) PW524 (D21S58) indicated with arrows.
Application of Chromoprobe M Multiprobe showed that the marker originated from chromosome 19. The presence of euchromatin in SMC(19) was confirmed using probes specific for D19S452, D19S269, D19S752, and D19S190. The alpha satellite D1Z7/D5Z2/D19Z3 probe showed a single hybridisation signal in all cells. Uniparental disomy of chromosome 19 was excluded using microsatellite polymorphism analysis at D19S535,APOC2C, and D19S246.
Case 4: 47,XX,+mar.ish der(20)(wcp20+, D20S1097−,PAX1−,GHRH−,D20Z1+)
The apparently monocentric marker chromosome was observed in all peripheral blood lymphocytes analysed. The mother's karyotype was normal and the father was not available for study. Following the suggestion of the clinician, probes specific for Y chromosome centromere were applied first in the FISH procedure giving negative results. Subsequently, probes for other chromosomes were used. Chromosome 20 centromeric probe gave positive results indicating the origin of the marker from this chromosome. None of the pericentromeric probes used revealed signals on SMC(20). The presence of the euchromatin could be shown only with the whole chromosome 20 paint (fig5). Biparental status of both homologues 20 was shown at four loci, D20S101, D20S85, PCK1, and D20S120.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) G banded chromosomes and (B) FISH with chromosome 20 library in case 4. The marker chromosome is shown by arrows.
Discussion
Among supernumerary marker chromosomes, those derived from non-acrocentric autosomes are rare and occur with a frequency of only ∼15%.26 Their clinical significance remains unclear and we are currently accumulating data which are needed to elucidate phenotype-karyotype correlations further.8 9 It is generally accepted that it is the presence of euchromatin that makes a marker chromosome deleterious to the phenotype. In this context, tiny markers composed mainly of heterochromatic centromere region are expected to be benign. However, the clinical consequences of small markers containing regions adjacent to centromeres are not clear. Our cases 2 and 4 belong to this category of markers and were found in phenotypically abnormal patients.
In case 2 with a der(6), developmental delay and dysmorphic features were found as was the case in four other published cases with SMC(6).9 21 23 However, no clear clinical phenotype can be found in these five mar(6) carriers. In two cases, the clinical picture may be complicated by the presence of the second marker derived from other chromosomes (9 and X) and in case 5 of Crollaet al 9 by the paternal isodisomy of normal 6 homologues.21 23 Moreover, in our case 2, two copies of mar(6) were found in almost half of the lymphocytes, which may influence the phenotype by the gene dosage mechanism.
The marker derived from chromosome 20 in case 4 was present in a female with abnormal gonads, primary amenorrhoea, hirsutism, and mild mental retardation. Anomalous gonads or hirsutism have not been found in any of the published mar(20) cases.7 9 23 27-29 The only common finding in all abnormal cases was growth retardation or delayed psychomotor development. An association between the clinical features, with the exception of mental retardation, and the presence of der(20) in our case seems problematical. We speculate that an additional copy of a novel sex differentiation gene could play a role in the anomalies described. Interestingly, Greenfield et al 30 mapped mouse Sryrelated Sox18 gene in a syntenic region of human chromosome 20q.
In cases 1 and 3, the marker chromosomes were bigger in size (∼28 Mb and ∼30 Mb, respectively) and had a ring structure. To the best of our knowledge, SMC(5) is the first marker identified derived from this chromosome. The clinical symptoms of its carrier excluding low frontal hairline, Baker's cysts, and talipes valgus match the previously described cases of partial trisomy 5p.31-35Interestingly, our data together with the dup(5) (p14p15.3) described by Zenger-Hain et al,34 could locate the strabismus trait to the 5p14 band.
In case 3, the marker derived from chromosome 19 was associated with partial duplication of 21q. As is the case in six of seven mar(19) reported to date (for review see Crolla8), our case was ascertained prenatally. In only two of these cases were abnormal phenotypes observed (cases 10 and 11 of Crolla et al 36). In the first one, mar(19) accompanied by another unidentified acentric marker chromosome was a variable ring and the abnormalities included large head, frontal bossing, hypotonia, epicanthic folds, and developmental delay. In the second case it was a minute marker in a child with failure to thrive and severe floppiness. In our patient only hypotonia was present; however, hypertelorism and microcephaly were described in trisomy 19q cases37-39 and agenesis of the corpus callosum was associated with trisomy 19q13.2-qter.40 The latter case of Crollaet al,36 together with our data, indicates that triplication of a putative gene located in chromosome band 19q13.2 may be responsible for agenesis of the corpus callosum. The symptoms observed in our proband, including hypotonia, hypertelorism, microcephaly, upward slanting palpebral fissures, and deep palmar and plantar creases, could account for partial trisomy of chromosome 21.41-45 The lack of a typical Down syndrome phenotype in our proband is in concordance with FISH results which showed that the DSCR is not triplicated. It is difficult to assess the contribution of trisomy 19p11p13.11 to the phenotype of our patient. To the best of our knowledge it has not been described previously.
In all our cases, as is generally accepted for markers containing euchromatic sequences, phenotypic abnormalities were observed. Although a causal relationship between an abnormal phenotype and the presence of the marker cannot be proven, the lack of such a relation seems rather unlikely. From these and published cases, no typical syndromes for mar(5), (6), (19), or (20) have yet emerged.
Acknowledgments
We are very grateful for the help of clinicians who provided patients for this studies: Dr Z Zabinski (case 1), Professor L Korniszewski (case 2), Dr J Pilch (case 3), and Dr M Szarras-Czapnik (case 4). The unique sequence probes were kindly provided by Dr Ung-Jin Kim from the California Institute of Technology in Pasadena, BAC clones CIT978SK-A-36F2, CIT987SK-A-277A9; Dr Mariano Rocchi from Istituto di Genetica, Università Degli Studi, Bari, Italy, DNA from YACs 882A10, 772B9, 819C1; Dr Christina Brahe from Istituto di Genetica Medica, Università Cattolica del Sacro Cuore, “Agostino Gemelli” in Rome, cosmid clone cosF; Dr Yusuke Nakamura from Laboratory of Molecular Medicine, Institute of Medical Science, University of Tokyo, DNA from cosmid clones cCI6-21, cCI6-28, cCI6-31, cCI6-46, cCI6-68, cCI6-84; Dr Harvey Mohrenweiser from Human Genome Center at Lawrence Livermore National Laboratory, cosmid clones 11994, 26418, 19977, 15694, 20120; Professor Philip Board from John Curtin School of Medical Research, Canberra, cosmid clone pJG1; Georges Lutfalla from UPR CNRS in Paris, DNA from cosmid clone PW524; Dr Jean M Delabar from Faculte Necker, Paris, BAC APP; Dr Mainrad Busslinger from Research Institute of Molecular Pathology, Vienna Biocenter, University of Vienna, DNA from cosmid clone Pax1-19; and Dr Kelly E Mayo from BMBCB, Northwestern University, Evanston, Illinois, DNA from cosmid clone HGRFcos49. We wish to thank Dr J Crolla for critical reading of the manuscript and Drs A Szpecht-Potocka and W Wiszniewski for helpful interpretation of microsatellite polymorphism results. The work was supported by the State Committee for Scientific Research, grant No 4 PO5E 036 11 and 6 P04 A070 14 and by a Herbert Quandt Stiftung der VARTA AG fellowship to Heike Starke.