Article Text

Abstract
We report a 13 year old boy with fragile X syndrome resulting from a de novo deletion of the FMR1 and FMR2 genes extending from (and including) DXS7536 proximally to FMR2 distally. The patient has severe developmental delay, epilepsy, and behavioural difficulties, including autistic features. He has epicanthic folds, in addition to facial features typical of fragile X syndrome, and marked joint hypermobility. We compare our patient to the three other cases reported in which both FMR1 and FMR2 are deleted. This case has the smallest deletion reported to date. All four patients have epilepsy and a more severe degree of mental retardation than is usual in fragile X syndrome resulting from FMR1 triplet repeat expansion. Three of the patients have joint laxity and two have epicanthic folds. We suggest that these features, in particular severe developmental delay and epilepsy, may form part of the characteristic phenotype resulting from deletion of both FMR1 and FMR2 genes. The diagnosis in this case was delayed because routine cytogenetics showed no abnormality and standard molecular tests for FMR1 triplet repeat expansion (PCR and Southern blotting) failed. Further DNA studies should be undertaken to investigate for a deletion where clinical suspicion of fragile X syndrome is strong and routine laboratory tests fail.
- fragile X
- FMR1
- FMR2
- deletion
Statistics from Altmetric.com
Fragile X syndrome is the commonest inherited cause of mental retardation, with a prevalence of between 1 in 4000 to 1 in 6000 males.1 2 Most cases are the result of amplification of a CGG trinucleotide repeat sequence located in the 5′ untranslated region of the FMR1 (FRAXA) gene on the long arm of the X chromosome. This sequence is unstable, a triplet repeat number of greater than approximately 200 causing inhibition of transcription of FMR1. Rarely, the clinical syndrome may result from deletion of all or part of the FMR1 (FRAXA) gene.3 In three cases reported previously, the FMR2 (FRAXE) gene lying just distal to FMR1 was also deleted.4-6 We describe a boy with a de novo deletion of the entire FMR1 and FMR2 genes and compare his features with those of the other reported cases.
Case report
The patient was born after an uneventful pregnancy by elective caesarian section for breech presentation. His birth weight was 4692 g and there were no perinatal problems. There was no relevant family history. Severe developmental delay became evident in the first year of life. He sat at 10 months and walked at 2 years 10 months. Now aged 13 years, he can speak in short sentences and has behavioural difficulties, with aggression and agitation. He also has autistic features including a dislike of change in routine and hand flapping. Since 2½ years he has had severe polymorphic epilepsy which has been difficult to control despite several anticonvulsant medications.
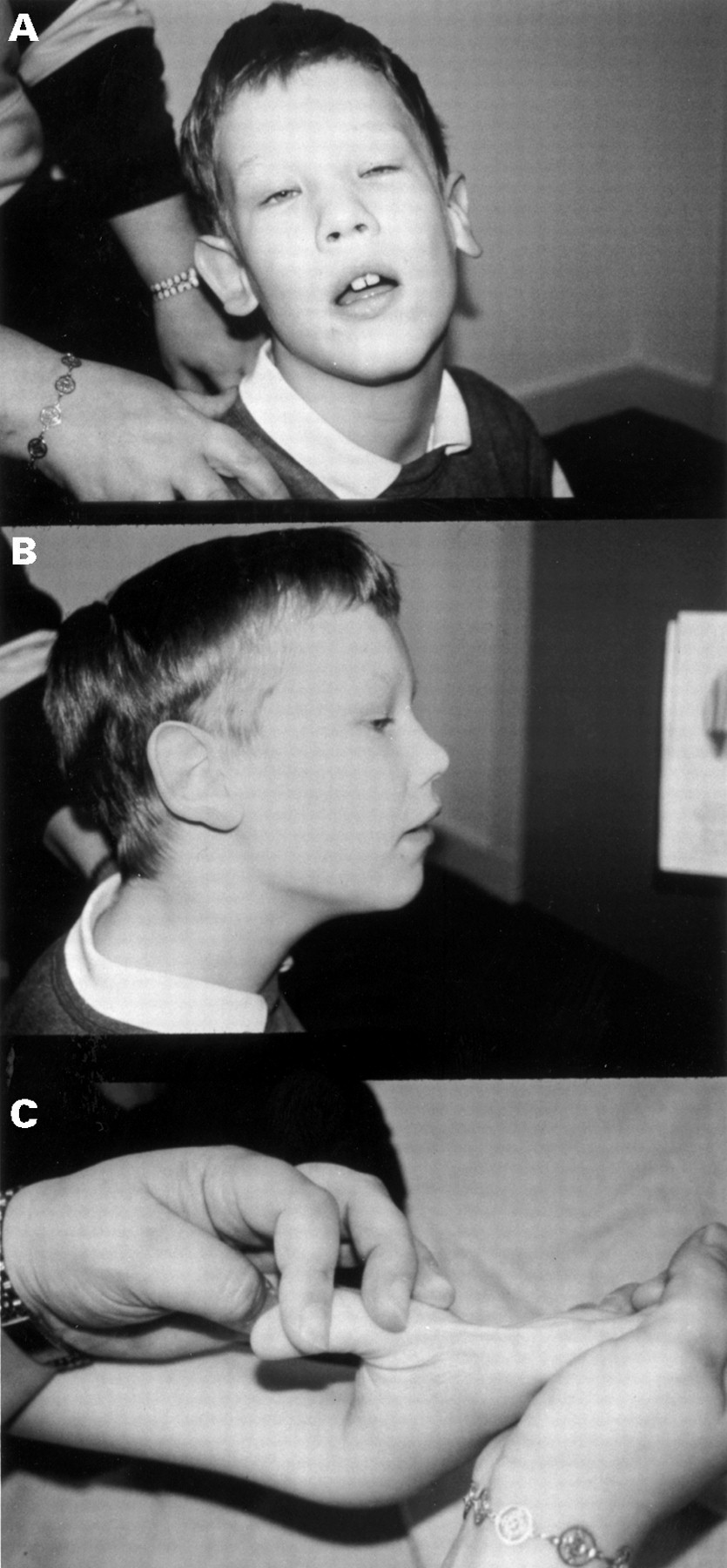
On examination, he has a large head (circumference above the 97th centile), a long face with prominent forehead and jaw, epicanthic folds, large ears, and marked joint hypermobility (fig 1). He also has a convergent squint.
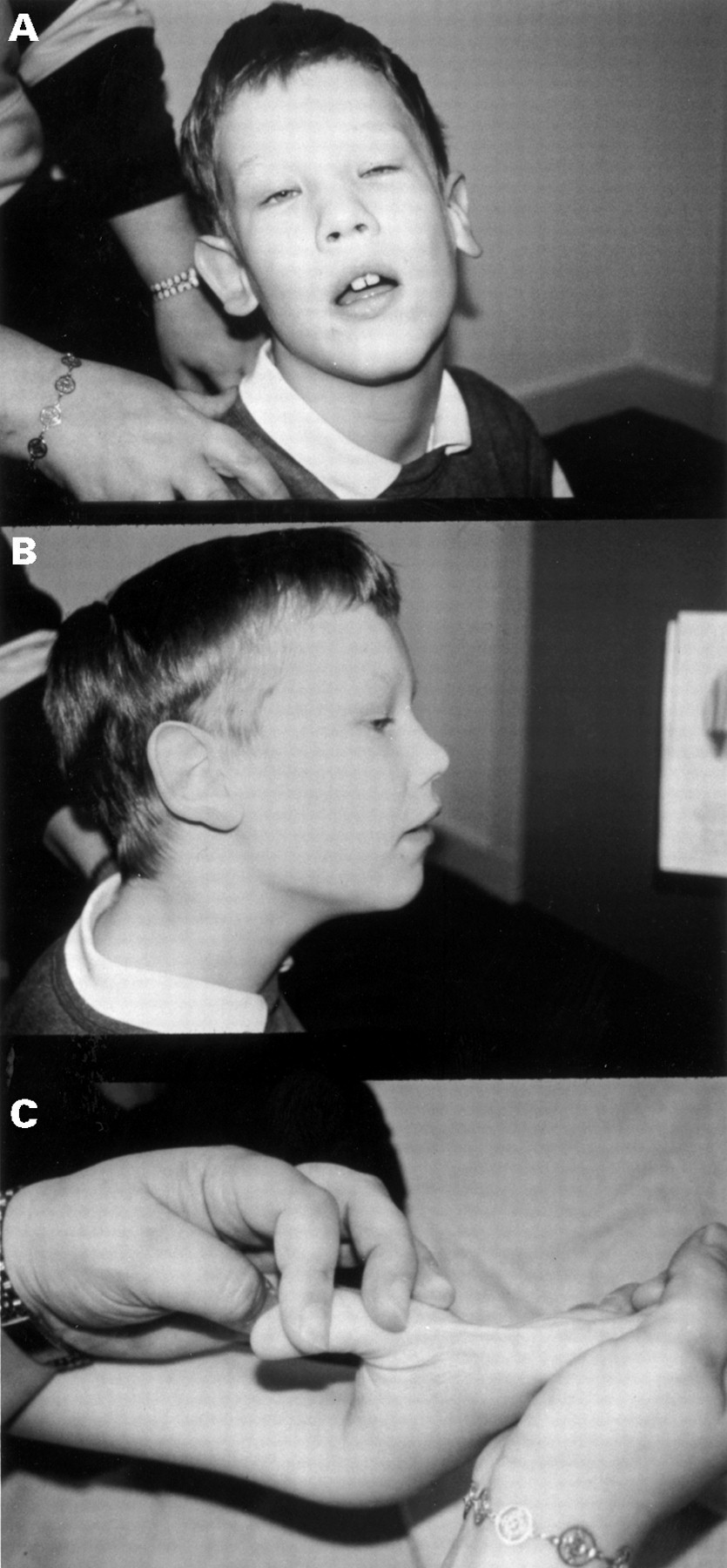
The proband. Note (A) prominent forehead and jaw and epicanthic folds, (B) prominent ears, and (C) marked joint laxity. (Photographs reproduced with permission.)
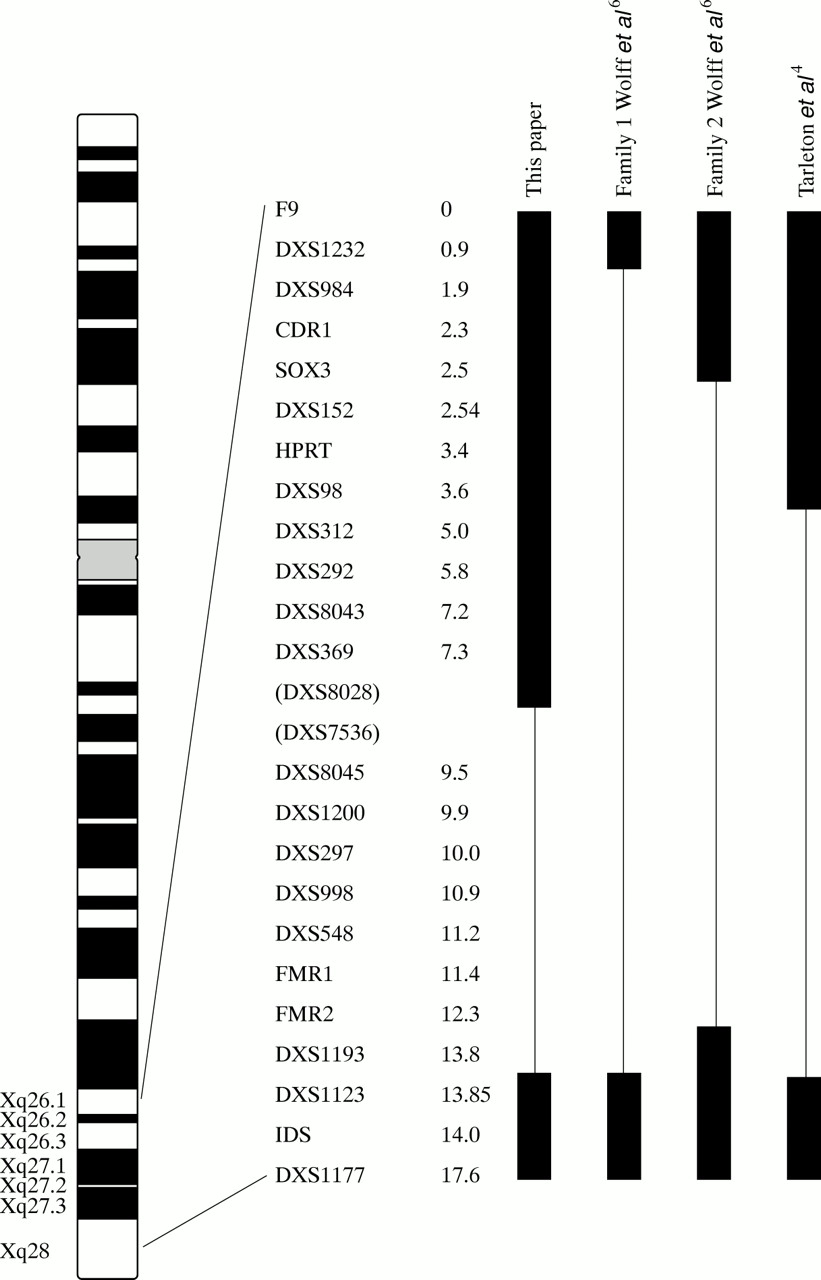
The karyotype was normal including culture in folate deficient medium. Routine molecular analysis by polymerase chain reaction (PCR) and Southern blotting using techniques designed to assess the size of the FMR1 triplet repeat region failed to produce a result. Further molecular investigation of regions flanking the gene showed that this was because of a de novo deletion of the entire FMR1 and FMR2 genes extending from (and including) DXS7536 proximally to FMR2 distally (fig2).
{kind=link}
{kind=link}
Map of the X chromosome in the region Xq26.1-Xq28 showing the markers deleted in this patient. Other deletions in this region are shown for comparison. The figures beside the markers represent physical distances from F9 in megabases (Mb) as reported in the Genetic Location Database (GLD) (http://cedar.genetics.soton.ac.uk). DXS7536 and DXS8028 are shown in brackets because although the GLD suggests that DXS8028 is distal to DXS7536, in our patient DXS7536 was deleted while DXS8028 was not, implying that the order of markers is as shown.
Discussion
Table 1 compares our patient with the other cases reported in which both FMR1 and FMR2 are deleted. He has the smallest deletion reported to date.
Comparison of clinical features in patients with deletion of FMR1 and FMR2
Epilepsy is seen in all four patients with deletions of both FMR1 and FMR2, who also have more severe intellectual impairment and a greater degree of joint laxity than is usual in patients with fragile X syndrome owing to FMR1 triplet repeat expansion. Our patient has epicanthus which is also seen in the case of Tarletonet al 4 and Albrightet al.5
In patients with deletion of FMR1 but intact FMR2, severe epilepsy and marked joint laxity are not common.3 These patients often have typical facial features, and the occurrence of atypical features such as eunuchoid habitus, cherubism, prominent occiput with double hair whorl, shawl scrotum, and anal atresia may be the result of deletion of other adjoining genes.
The FMR2 associated phenotype is said to be milder than that caused by loss of expression of FMR1. However, losing both FMR1 and FMR2 genes appears to cause a more severe disorder of brain development. The clinical features shared by our patient and those previously reported with FMR1 and FMR2 deletions, in particular moderate to severe developmental delay and epilepsy, suggest that these may be characteristic features which result from loss of both these genes. Further case reports will help to define this phenotype more clearly.
Finally, it should be noted that the diagnosis in our patient was delayed because routine laboratory investigations were negative (cytogenetics) or failed (DNA). Further investigations of DNA from the child and his mother indicated that the “failure” of the routine DNA test was the result of a de novo deletion in the child. It is important to be aware of this mechanism and to undertake further DNA studies if there is a strong clinical suspicion of fragile X syndrome.