Article Text

Abstract
Angelman syndrome (AS) is a neurological disorder with a heterogeneous genetic aetiology. It most frequently results from a de novo interstitial deletion in the 15q11-q13 region, but in a few cases it is caused by paternal uniparental disomy (UPD) or an imprinting mutation. The remaining 20 to 30% of AS patients exhibit biparental inheritance and a normal pattern of allelic methylation in the 15q11-q13 region. In this latter group, mutations in the UBE3A gene have recently been shown to be a cause of AS. Here we describe the phenotypic expression in 14 AS cases involving eight UBE3A mutations. These comprise 11 familial cases from five families and three sporadic cases. Subtle differences from the typical phenotype of AS were found. Consistent manifestations were psychomotor delay, a happy disposition, a hyperexcitable personality, EEG abnormalities, and mental retardation with severe speech impairment. The other main manifestations of AS, ataxia, epilepsy, and microcephaly, were either milder or absent in various combinations among the patients. In addition, myoclonus of cortical origin was frequently observed with severe fits inducing myoclonic seizures. The majority of the patients were overweight. This study showed that ataxia, myoclonus, EEG abnormalities, speech impairment, characteristic behavioural phenotype, and abnormal head circumference are attributable to a deficiency in the maternally inherited UBE3A allele. Furthermore, analysis of mutation transmission showed an unexpectedly high rate of somatic mosaicism in normal carriers. These data have important consequences for genetic counselling.
- Angelman syndrome
- cortical myoclonus
- UBE3A mutation
- somatic mosaicism
Statistics from Altmetric.com
In 1965, Angelman1 described three unrelated children with severe mental retardation, ataxic movement, a happy demeanour with inappropriate paroxysms of laughter, absence of speech, seizures, and microcephaly with a characteristic facial appearance. Since then, Angelman syndrome (AS) has been more frequently recognised and its prevalence is estimated to be about 1/20 000 live births.2 3 The majority of AS cases are sporadic. Nevertheless, several reports have shown that AS may recur within a family4 and the incidence of familial AS cases has been estimated to be 7% in two extensive surveys.5 6
AS is caused by different genetic alterations involving the imprinted chromosome 15q11-q13 region.7 This region also contains the Prader-Willi syndrome (PWS) locus. AS results from the lack of contribution of normally active maternally inherited gene(s) on chromosome 15q11-q13, whereas PWS results from the lack of contribution of normally active paternally inherited genes. Familial studies have suggested that AS could result from a single gene mutation, with phenotypic expression only after maternal transmission.8Recently, a gene encoding a ubiquitin-protein ligase (UBE3A), previously mapped within the AS critical region, has been found to be mutated in several AS patients.9 10 Furthermore, UBE3A is expressed predominantly from the maternal allele in brain.11 On the basis of molecular and cytogenetic criteria, six distinct mechanisms have been shown to cause AS.12 It most frequently results from a maternal de novo interstitial deletion in the 15q11-q13 region (class 1), but rare complex structural chromosome rearrangements involving the 15q11-q13 region have been reported (class 2). In a few cases, it is caused by paternal uniparental disomy (UPD) of chromosome 15 (class 3), or an imprinting control centre mutation identified by abnormal methylation pattern (IM) (class 4). Approximately 20 to 25% of AS patients exhibit biparental inheritance and a normal pattern of allelic methylation in the 15q11-q13 region. In this group, UBE3A mutations have been found in 20% of patients (class 5). The remaining cases, however, have no identifiable molecular abnormality, and the diagnosis of AS is on clinical grounds (class 6).
Between 1989 and 1997, we clinically diagnosed 59 AS cases from 52 different families in the Department of Medical Genetics in Marseilles. This survey included six families with more than one AS child. A group of 21 AS patients from 14 different families, including seven sporadic cases and the six familial cases, showed biparental inheritance and a normal pattern of allelic methylation in the 15q11-q13 region. Recently, UBE3A mutations have been identified in 14 patients from eight unrelated families.13 They comprised 11 familial cases from five families and three sporadic patients. In the familial cases, there were five distinct mutations consisting of two insertion mutations (exon 12 and exon 16) and two deletion mutations (exon 9), which cause premature termination of the predicted open reading frame, and one insertion mutation (exon 15) that results in the insertion of an isoleucine at position 802. In the sporadic cases, two distinct insertion mutations (exon 8 and exon 16) and one insertion deletion mutation (exon 10) were found. Extensive clinical studies of sporadic14-16 or familial AS patients4 were reported before the recognition of UBE3A and imprinting mutations. We describe the clinical features of these AS patients and the transmission of the mutation in the families.
Subjects and methods
The 14 patients, comprising 11 familial cases from five families and three sporadic patients, were referred to us for unexplained developmental delay between 1989 and 1995. The same geneticist and neuropaediatrician made the clinical evaluation of all patients. Clinical history, anthropometric data, physical and neurological findings, EEG studies, and extensive investigation of the maternal pedigree were registered using our standard protocol adapted from Angelman1 and Robb et al.17 Clinical diagnosis of the patients was made before the DNA results were available.
Molecular and cytogenetic studies showed biparental inheritance of chromosome 15 with a normal methylation pattern (loci D15S63 and SNRPN-exon 1) in the 15q11-q13 region in all probands. Cytogenetic analysis, DNA extraction, methylation analysis, and microsatellite studies were performed following standard protocols.18 To study the segregation of the 15q11-q13 haplotypes, DNA samples were collected from nuclear families with both affected and unaffected offspring, as well as members of the maternal family. Patients and their relatives were genotyped using microsatellites for loci within the AS/PWS region: p4.3R, D15S128 (Généthon), D15S210 (Généthon), D15S113, GABRB3-CA, 155.1CA, and 85.CA.
Mutation analysis involving UBE3A was performed by SSCP analysis and DNA sequencing and is reported in detail by Malzacet al.13 Mutation results are presented in table 1. Transmission of the mutations was studied by SSCP analysis and DNA sequencing.
Clinical manifestations of 14 patients from eight families resulting from UBE3A mutation
Results
FAMILY PEDIGREES
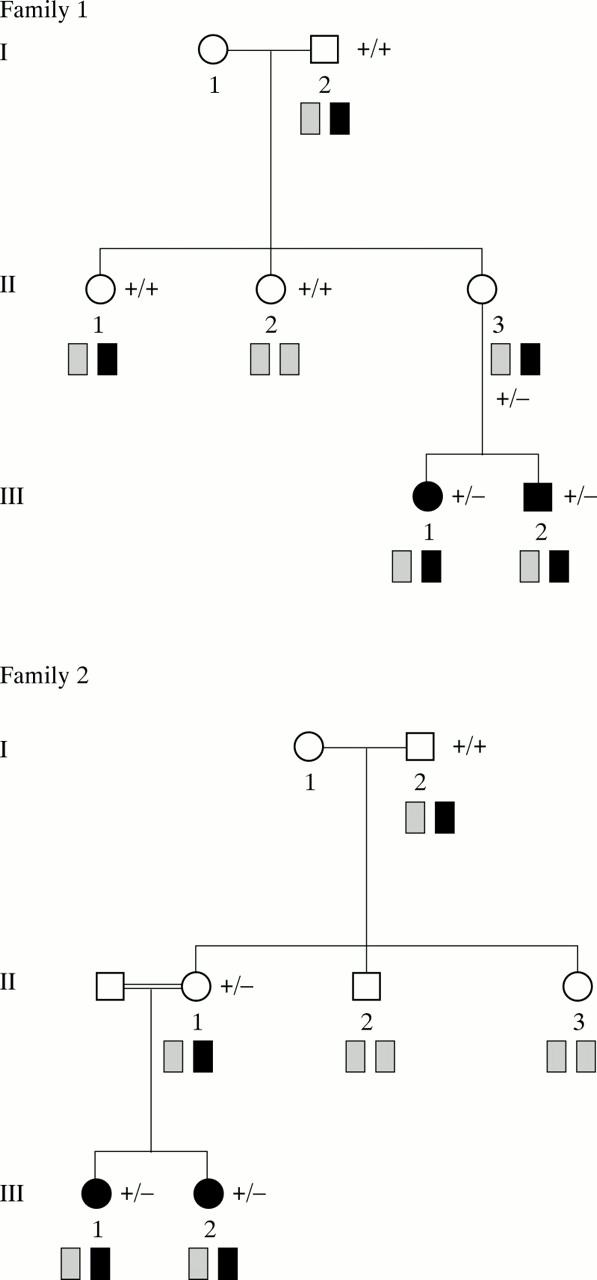
Families 1 and 2 are illustrated in fig 1A. The sibs comprised one male and one female (III.1 and III.2) in family 1 and two females (III.1 and III.2) of consanguineous parents in family 2. The pedigree of family 3 is shown in fig 1B. Two phenotypically normal sisters (III.5 and III.7) and a paternal aunt (II.1) produced, respectively, two, one, and one offspring with AS syndrome, two of whom were under evaluation (IV.8 and IV.14). Before the family was referred to us, one patient (IV.9) died aged 30 years and III.1 died at the age of 70 years. The medical records and photographs confirmed that they had both suffered from AS. The pedigree of family 4 is illustrated in fig 1B. Two phenotypically normal sisters (II.2 and II.3) produced, respectively, one and two offspring with AS (III.2; III.3 and III.4). The pedigree of family 5 is illustrated in fig 1B. The female patient (V.2) was the first to be diagnosed with AS. Her mother spoke of a male relative with unexplained mental retardation, living a long way from France. The mother of this subject (V.10) was her paternal first cousin. Medical records and photographs were obtained through correspondence with the parents, confirming the diagnosis of AS. An extensive investigation of maternal relatives eventually showed that seven AS cases had occurred in several sibships over five generations. Of the sporadic cases, two were only children (cases 1 (III.1) and 2 (III.1), fig 1C). Sporadic case 3 (III.3) has an unaffected brother and sister (fig 1C). The maternal chromosome 15 haplotype for the affected AS patients was determined using microsatellites for loci within the PWS/AS region. The maternal AS related haplotype is shown in black and the normal chromosome 15 by hatching. Segregation of the AS related haplotype is shown for each family. UBE3A mutations are denoted by (−) and the normal allele by (+). Transmission of the mutation is shown for affected and unaffected subjects who were tested (mos=somatic mosaicism).



(A-C) Pedigrees of eight families including five families with recurrence of AS and three sporadic cases.
TRANSMISSION OF THE MUTATION
In the familial cases, segregation analysis of chromosome 15 showed that the affected children had inherited the same maternal chromosome 15. The grandpaternal origin of the AS related haplotype was shown in families 1, 2, 4, and 5 (fig 1) (in family 3, the grandfather’s DNA was not available). This result was consistent with dominant transmission of a mutation at an imprinted locus,8 and potential unaffected female carriers were identified in all these families. Our study of UBE3A mutation transmission confirmed these results. In addition, in family 4, we found somatic mosaicism of the UBE3A mutation in the phenotypically normal grandfather by SSCP analysis (fig2).

SSCP results in family 4. SSCP analysis was performed using primers E63I/E63J (exon 9). The lanes include two AS probands (lane 1: III.3, lane 2: III.2), their phenotypically normal mothers (lane 3: II.3, lane 4: II.2), their aunt (lane 5: II.1), and their maternal grandfather (lane 6: I.2) (see reference number of each subject on the family pedigree). A normal control was included (lane 7). II.2 (lane 5) and the normal control (lane 7) have four normal bands. The two AS probands (lanes 1 and 2), their mothers (lanes 3 and 4), and maternal grandfather (lane 6) have an additional lower (mutant) band. All have normal and mutant bands of equal intensity except the grandfather who has a lower band of <5% of the intensity of the upper band, indicating mosaicism for the UBE3A mutation (the autoradiograph was overexposed to visualise the mutant band in the grandfather).
In the three sporadic cases, the maternal chromosome 15 inherited by the affected patients was grandpaternal in origin. Segregation analysis of the AS related haplotype showed that this haplotype was shared with an unaffected sib in case 3 and three unaffected first cousins in case 1 (fig 1). These findings suggested either that a de novo mutation had arisen in the affected child or his mother or that germline mosaicism was present in the mother. The study of the transmission of a known mutation confirmed the results of segregation analysis. In addition, in case 1, an only child, we found somatic mosaicism in his phenotypically normal mother by SSCP analysis. In case 2, we are able to conclude that the mother’s sister (II.1) who shared the AS related haplotype was not at risk of being a carrier.
NATURAL HISTORY AND CLINICAL MANIFESTATIONS
Data of the patients are shown in table 1. The patients comprised six females and eight males aged from 9 months to 33 years at diagnosis.
Pregnancy and neonatal period
The mother’s age at the birth of the AS patient ranged from 22 to 32 years (mean 28 years) and the father’s age at birth ranged from 23 to 34 years (mean 28.7 years). Pregnancies, anthropometrics at birth, and head circumferences were normal or within the normal range in all cases. Feeding difficulties were reported in three cases and included difficulty with sucking (one case) and frequent vomiting (two cases).
Neurobehavioural findings
Motor development was delayed in all patients. The mean age of onset of walking was 33 months, with 10 patients walking by 3 years of age. None of the adults had loss of walking. Ataxia was typical in three cases, mild in three cases, and moderate in eight cases. Seizures were reported in 11 of 14 cases (78.6 %). The age of onset of seizures ranged from 6 months to 20 years (mean age 5.4 years). In 8 of 11 patients, only one episode of seizures was noticed in a long follow up of 5 to 10 years. The most frequent ictal patterns were atypical absences and myoclonic seizures. Episodes of seizures were easy to control with treatment in all patients. The three patients unaffected by seizures were 7, 20, and 30 years of age, respectively. However, the available EEG studies performed in childhood showed characteristic AS EEG patterns in all cases, either with or without seizures.19 In some adults, improvement in the EEG pattern was observed, as previously described by Boydet al.19 Myoclonus of cortical origin was observed in 13 of 14 patients.20 In one patient (family 4, patient III.3), myoclonus was not evident clinically and was only confirmed using back averaging electroencephalography. Severe episodic fits of myoclonus, triggered by stress, obviously induced the onset (family 4, III.2 at 20 years), or the recurrence of myoclonic seizures (sporadic case 3, at 15 years, after a seizure free period of seven years; family 3, IV.14; family 5, V.10). Sleep disturbance was recorded in 10/14 cases and persisted into adulthood in two patients. All patients had severe speech impairment. Half of the patients had acquired between four and 10 words, but they were only able to utter one or two syllables of each word. Nevertheless, all of them had developed a highly efficient system of non-verbal communication and they showed a level of comprehension better than that generally reported in typical AS cases. They all had a happy disposition, moderate hyperexcitability, and appropriate laughter and hand flapping when excited.
Physical findings
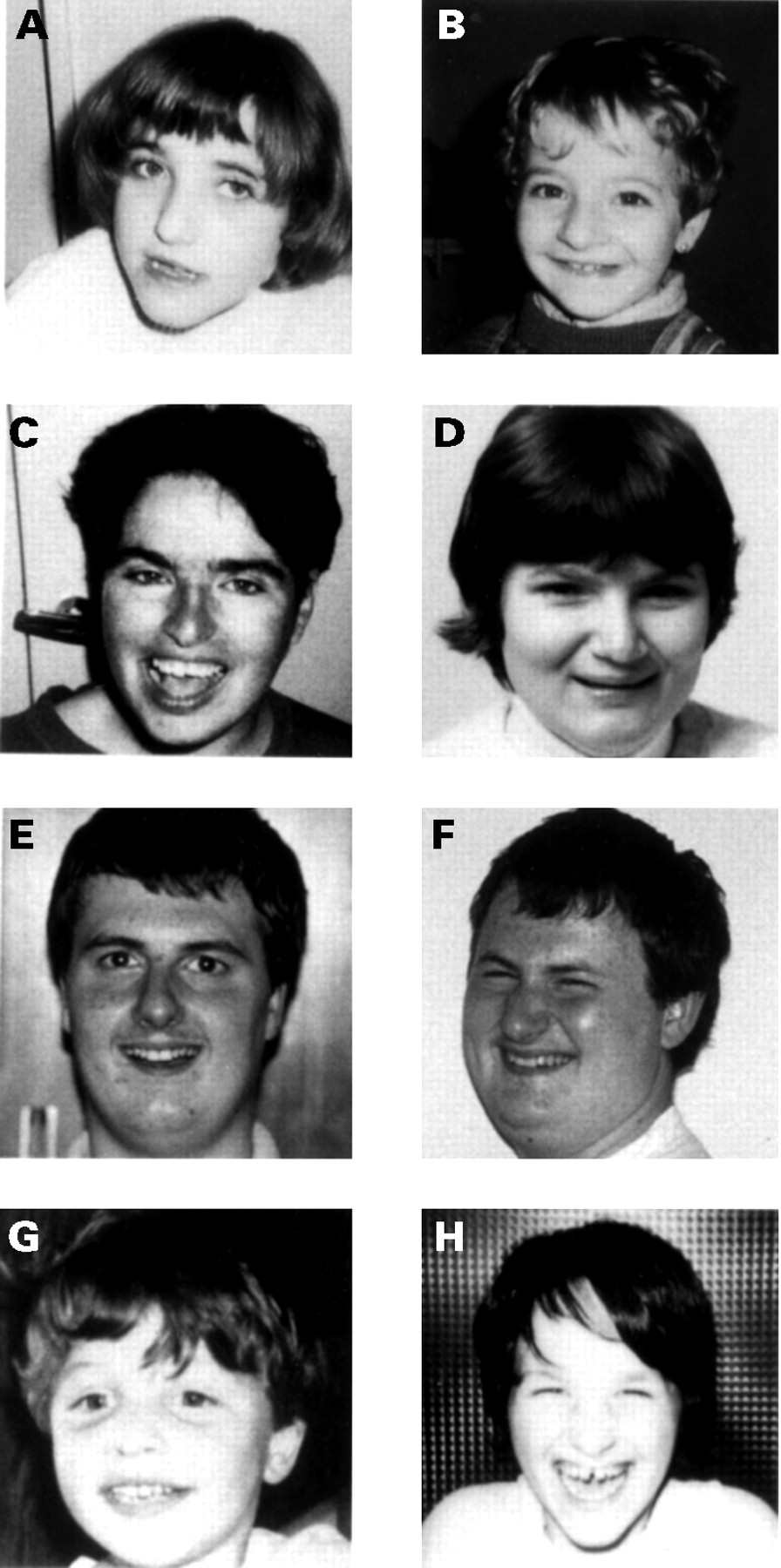
A delayed increase in head circumference was observed in 13 patients (93%), resulting in absolute microcephaly (equal to −2 SD) in six cases (42.8%) and relative microcephaly (between −1.5 SD and −1 SD) in seven cases (53.8%). One male patient (family 5) had a head circumference of average size, whereas his female cousin was microcephalic. Facial dysmorphism was mild in nine patients (families 1, 3, and 4, and sporadic cases 1 and 2). The characteristic large mouth and prognathism were not observed in these patients (fig 3). Strabismus was observed in three cases and facial asymmetry in two cases. None was hypopigmented. Scoliosis was present in two of eight adult or adolescent cases. Growth retardation (−2 SD) was observed in only two female adult cases (14.3%), whereas the affected males (families 3 and 5) were of normal height. In these two families, unaffected female carriers were short. Obesity (⩾2 SD relative to height) with hyperphagia was present in seven of 14 cases (64.3%).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Facial dysmorphism of eight patients with UBE3A mutations. (A, B) Two affected sisters. (C, D) Two adult females, one from family 5 (C) and the other from family 4 (D). (E, F) Mild facial dysmorphism of two brothers from family 4. (G, H) Two sporadic cases, one with mild dysmorphism and the other with the characteristic facial dysmorphism.
Discussion
The diagnosis of AS in patients with biparental inheritance and a normal methylation pattern is essentially based on clinical findings. The typical phenotype of AS comprises severe mental retardation, epilepsy, lack of speech, ataxic movements, outbursts of inappropriate laugher, large mouth and chin, microcephaly, and an abnormal EEG. The clinical diagnosis of AS is known to be difficult, particularly at an early age21 but also in mentally retarded adults,16 because other clinical disorders can mimic the features of AS. These conditions include Rett syndrome, α thalassaemia-X linked mental retardation, autism, and ataxic cerebral palsy. With reference to published familial AS cases, no UBE3A mutation was found in one family with two affected brothers.9 22Because of a recombination event previously described in this family, Fang et al 23 recently suggested that the mutation is towards the 5′ end of the UBE3A locus. An alternative explanation is that they do not have AS, but have α thalassaemia-X linked mental retardation syndrome. This is supported by the clinical manifestations of severe hypotonia and mental retardation, characteristic facial dysmorphism, and genital anomalies.
In the present study, we provide information on the natural history and clinical findings of 14 patients with UBE3A mutations. To the best of our knowledge, only one report of two AS patients with UBE3A has been published with a detailed clinical description.24 One patient had a typical phenotype and the other an atypical phenotype, in that she has less severe ataxia, no inappropriate laughing or epilepsy, and her EEG was normal at 2 years. In the present study, the affected members of family 5 and the sporadic case 3 showed the most typical AS phenotype,21 all the others exhibiting a slightly atypical one. In these cases, we made the clinical diagnosis on the combination of psychomotor delay, happy disposition, a hyperexcitable personality, EEG abnormalities, and mental retardation with severe speech impairment. The other main manifestations of AS, ataxia, epilepsy, and microcephaly, were either mild or absent in various combinations among the patients (table 1). In addition, we observed intrafamilial variability of clinical manifestations in four families (table 1, families 2, 3, 4, and 5). At an early age, the behavioural phenotype of happy disposition and hyperexcitability is probably the most important manifestation and appears to be decisive in the differential diagnosis of patients with psychomotor and language delay. The characteristic EEG abnormalities described by Boyd et al 19 are also particularly important and may be a good diagnostic clue. This study shows, however, that they are not always present at an early age (table 1), as also found by Funget al 24 in one case.
The abnormal motor pattern in AS is complex and usually described as jerky, tremulous, or dystonic. Guerrini et al 20 reported that this particular motor pattern is related to myoclonus of cortical origin. For the first time, we show that myoclonus is effectively a consistent manifestation of patients with a UBE3A mutation. It was found in all the patients except the youngest. However, myoclonus is not evident clinically in all patients, but can be shown using back averaging electroencephalography (patient III.3, family 4). This finding suggests that myoclonus results from loss of the maternal UBE3A allele and not GABRB3 as proposed by Guerrini et al.20 In addition, severe episodes of myoclonus were obviously correlated with myoclonic seizures in four patients. Piracetam treatment reduced myoclonus in some patients. The treatment appeared to prevent the occurrence (family 4, III.4) or recurrence of seizures (sporadic case 3). These observations provide useful information for specific management of AS patients. Finally, another finding is the tendency to become overweight, as has previously been described in AS patients.14
Most of the UBE3A mutations reported here appear to cause premature chain termination. The only exception is the mutation found in family 5, which causes a one amino acid insertion. The structure of UBE3A protein has a highly conserved hect domain encoded by exons 9-16.25 One might expect that a mutation in exon 9 would be more severe than one in exon 15 or 16. Our study does not support this, since no significant clinical difference was found between the patients with truncating mutations in exon 9 (families 1 and 4) or exon 16 (family 3, sporadic case 2).
The present study shows that a milder than usual AS phenotype is associated with UBE3A mutations.21 The phenotype in mutant mice with maternal deficiency of Ube3a can be compared with AS. Interestingly, milder ataxia and inducible seizures were observed in these mutant mice.26 Given the difficulty of both clinical diagnosis of AS and molecular analysis of UBE3A, we would suggest that the consistent manifestations found here (psychomotor delay, happy disposition, hyperexcitable personality, EEG abnormalities, mental retardation, and severe speech impairment) should be the minimal clinical criteria required before screening for UBE3A mutations.
In our series of 21 AS patients from 14 families with biparental inheritance of chromosome 15 and a normal methylation pattern, the rate of UBE3A mutation was 57% (83% in familial and 42.8% in sporadic cases). This rate is consistent with the recent study reported by Fanget al.23 In our survey of AS, UBE3A mutation appeared to be the second most frequent cause of AS (unpublished data). This is an important result because of the mode of inheritance. Previous analysis of several different AS families27 28 and one large pedigree29showed autosomal dominant transmission with phenotypic expression dependent on parental imprinting. This mode of inheritance leads to a high risk of recurrence (50%) for unaffected female carriers. Family 5 illustrates the necessity for extensive research into the maternal pedigree of affected subjects and females at risk in order to provide an accurate assessment of the risk of recurrence. Until now, because of this mode of inheritance, the risk of recurrence in sporadic cases was estimated empirically to be high for the mother of an affected child and the female relatives sharing the same chromosome 15 haplotype. With the identification of a mutation in UBE3A as a cause of AS, genetic counselling can become more accurate in familial cases and is now possible in some sporadic cases. However, we found somatic mosaicism in two of eight families, with one case occurring in a grandfather and another in the mother of an affected only child. In the latter case, an accurate risk of recurrence is difficult to establish, but it should be considered high. Six of the eight mutations found (75%) are maternally inherited and two appear to be de novo, but we could not exclude the possibility of germline mosaicism. The high rate of somatic mosaicism found suggests that germline mosaicism may be frequent. The risk of recurrence in cases of de novo mutation should be given with great caution.
Acknowledgments
We are grateful to all the families and the clinicians who referred patients for their cooperation in this study. We particularly thank Professor M Lalande and Dr J Wagstaff for their help in screening for the UBE3A gene in our patients.