Article Text

Abstract
Familial hypertrophic cardiomyopathy is a genetically heterogeneous autosomal dominant disease, caused by mutations in several sarcomeric protein genes. So far, seven genes have been shown to be associated with the disease with the β-myosin heavy chain (MYH7) and the cardiac myosin binding protein C (MYBPC3) genes being the most frequently involved.
We performed electrocardiography (ECG) and echocardiography in 15 subjects with hypertrophic cardiomyopathy from a French Caribbean family. Genetic analyses were performed on genomic DNA by haplotype analysis with microsatellite markers at each locus involved and mutation screening by single strand conformation polymorphism analysis. Based on ECG and echocardiography, eight subjects were affected and presented a classical phenotype of hypertrophic cardiomyopathy. Two new mutations cosegregating with the disease were found, one located in the MYH7 gene exon 15 (Glu483Lys) and the other in the MYBPC3 gene exon 30 (Glu1096 termination codon). Four affected subjects carried the MYH7 gene mutation, two the MYBPC3 gene mutation, and two were doubly heterozygous for the two mutations. The doubly heterozygous patients exhibited marked left ventricular hypertrophy, which was significantly greater than in the other affected subjects.
We report for the first time the simultaneous presence of two pathological mutations in two different genes in the context of familial hypertrophic cardiomyopathy. This double heterozygosity is not lethal but is associated with a more severe phenotype.
- hypertrophic cardiomyopathy
- genetics
- β-myosin heavy chain gene
- cardiac myosin binding protein C gene
Statistics from Altmetric.com
Familial hypertrophic cardiomyopathy (FHC) is an autosomal dominant disease characterised by left ventricular hypertrophy (LVH) and myofibrillar disarray.1 2 It is genetically heterogeneous and can be caused by mutations in the β-myosin heavy chain gene (MYH7)3 located on chromosome 14, the cardiac troponin T gene (TNNT2) on chromosome 1, the α-tropomyosin gene (TPM1)4 on chromosome 15, the cardiac myosin binding protein C gene (MYBPC3)5 6 on chromosome 11, the ventricular myosin essential (MYL3) and regulatory (MYL2) light chain genes7 respectively located on chromosomes 3 and 12, and the cardiac troponin I gene (TNNI3)8 located on chromosome 19. All these genes encode for proteins involved in the sarcomeric function, so hypertrophy might be the result of a compensatory phenomenon to quantitative or qualitative abnormalities of the contractile process. The pattern and degree of LVH show considerable variability, as does the clinical expression, ranging from no symptoms to heart failure and premature sudden death.1 In our experience, in about one third of families, the FHC results from mutations in MYH7 and another third from mutations in MYBPC3. We report here the genetic and clinical data of a French Caribbean family with hypertrophic cardiomyopathy, which was first excluded from the chromosome 14, 1, 15, 11, and 12 loci by haplotype linkage analyses. A systematic screening of mutations in MYH7 and MYBPC3 allowed us to identify the segregation of two mutations, one in MYH7 and the other in MYBPC3, and we analysed the phenotype-genotype relationship.
Methods
SUBJECTS
After informed consent was obtained by applying the guidelines of the Comité d’Ethique du Centre Hospitalier Universitaire de la Pitié-Salpêtrière (Paris), all genotyped subjects underwent clinical and cardiovascular examination including a 12 lead electrocardiogram (ECG) and M mode, two dimensional, and Doppler echocardiography at the time of genotyping. The diagnosis of FHC was based on ECG and echocardiography as previously described9: briefly, major echo diagnostic criteria were defined by a left ventricular end diastolic maximal wall thickness >13 mm or the presence of major ECG abnormalities (LVH assessed by a Romhilt-Estes score ⩾4 and/or Q waves >0.04 sec or >1/3R wave and/or significant ST-T changes). End diastolic left ventricular wall thickness measurements were obtained at different locations (anterior and posterior septum, lateral and posterior wall) from the parasternal short axis view, at both the mitral valve and the papillary muscle levels, and at the parasternal long axis view. The degree and extent of left ventricular hypertrophy were assessed by the maximal wall thickness and the scoring system proposed by Spirito and Maron.10 The distribution of hypertrophy was assessed according to the classification of Maron et al.11
GENETIC ANALYSIS
Haplotype analyses were performed on different loci with microsatellite markers provided by the Généthon human genetic linkage map.12 Linkage to chromosome 14 was carried out with two MYH7 intragenic microsatellites (MYOI and MYOII), linkage to chromosome 1 with three extragenic markers (D1S306, D1S456, and D1S249), linkage to chromosome 15 with two extragenic and one intragenic microsatellite markers (D15S153, HTMα(CΑ), and D15S131), linkage to chromosome 11 with four extragenic markers (D11S4133, D11S1344, D11S1350, and D11S1326), and linkage to chromosome 12 with six extragenic markers (D12S84, D12S1645, D12S1339, D12S1344, D12S1583, and D12S354).13
Screening of mutations was performed after amplification of each MYH7 and MYBPC3 exon with oligonucleotide primers determined in the flanking intronic sequences and single strand conformation analysis as previously described.6 14
Then, the PCR products were sequenced on both strands according to the method of Sanger et al 15 using rhodamine fluorescent dideoxynucleotides. The reaction products were then electrophoresed on an automated laser fluorescent DNA sequencer (Applied Biosystems).
STATISTICAL ANALYSIS
Values were expressed as mean (standard deviation (SD)); comparison between groups was performed with the Mann-Whitney test. Differences were considered significant at p<0.05.
Results
The pedigree of the family is shown in fig 1. Fifteen subjects were studied and eight were clinically affected. Clinical and echocardiographic characteristics of affected subjects are shown in table 1. All clinically affected subjects were adult and had a maximal left ventricular end diastolic wall thickness >13 mm. None had isolated major ECG abnormalities and none had significant intraventricular obstruction.
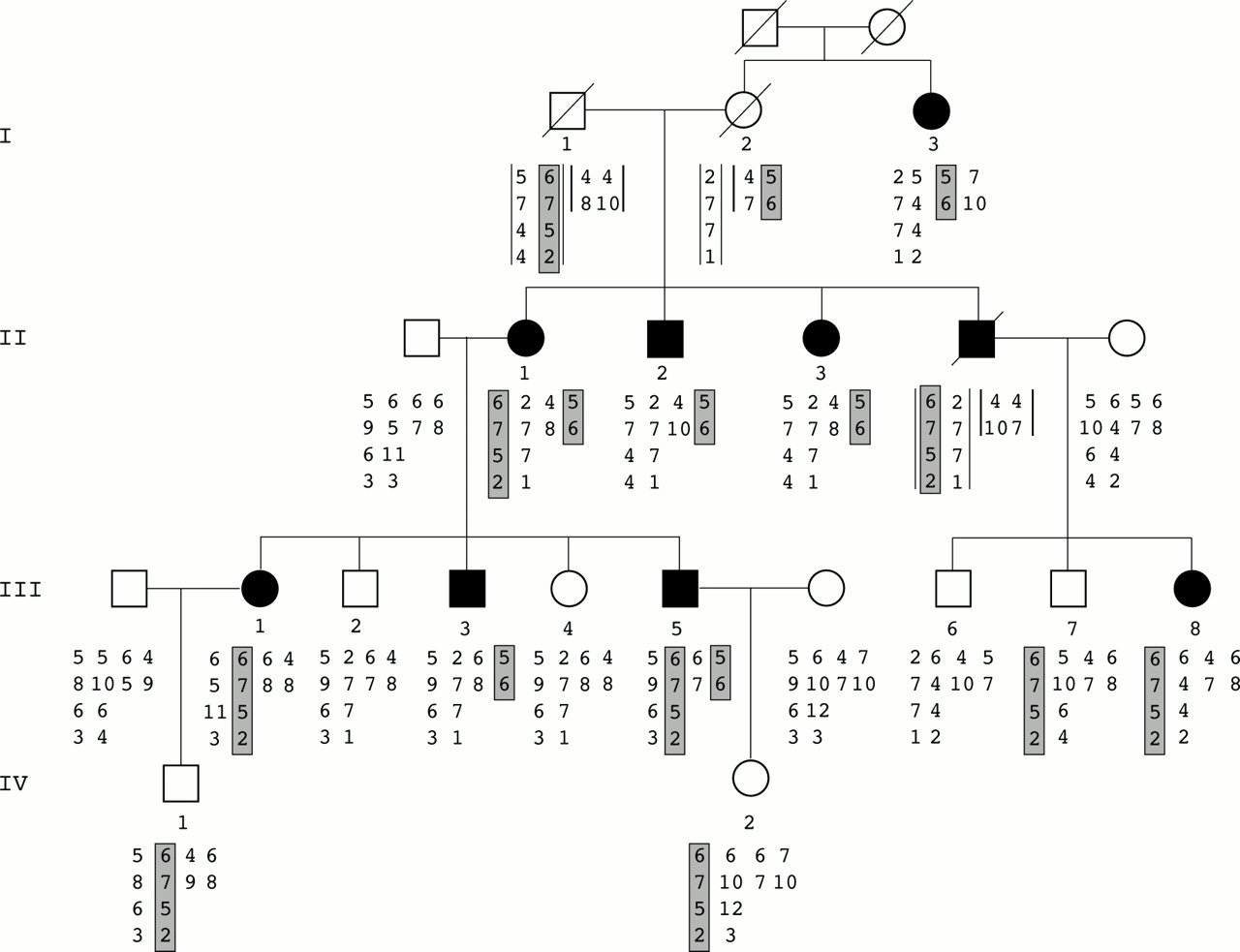
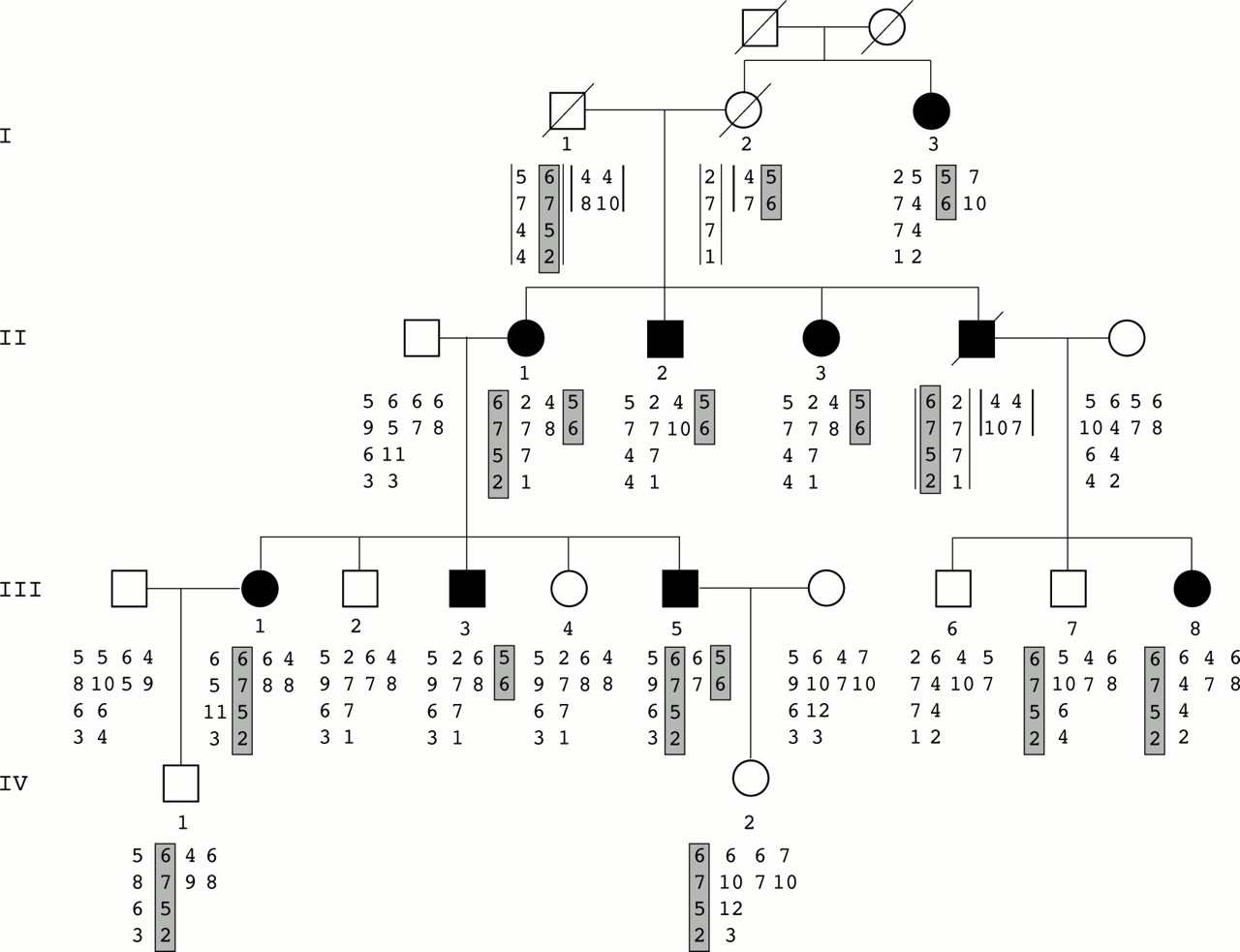{kind=link}
Inheritance of familial hypertrophic cardiomyopathy and haplotype analysis. Alleles shown below each family member are listed from top to bottom. On the left, chromosome 11 markers are indicated (D11S4133, D11S1344, D11S1350, D11S1326) and on the right the two MYOI, MYOII markers of chromosome 14. Data in brackets were deduced. Affected haplotypes are shaded in grey, 6-7-5-2 for the chromosome 11 locus and 5-6 for the chromosome 14 locus.
Clinical, ECG, and echocardiographic characteristics of affected subjects
Preliminary haplotype analyses showed no linkage with any of the loci tested. This finding suggested the presence of an unknown locus. Despite this possibility, we performed a mutation detection analysis of the 35 MYBPC3 exons in the proband (II.1). This analysis by SSCP showed an abnormal profile in exon 30, and sequence analysis subsequently showed a G to T mutation at codon 1096 leading to a TAA termination codon. Analysis of all the family members showed that only the clinically affected subjects III.1, III.5, and III.8 carried this mutation. Microsatellite marker analysis at the MYBPC3 locus showed that the haplotype 6-7-5-2 cosegregated with the mutation (fig 1) but seemed to be transmitted by the dead subject I.1. The finding that subject I.3 was clinically affected but did not bear either the affected haplotype or the MYBPC3 mutation led us to postulate that another mutation could be involved in the family. Therefore we analysed the other locus haplotype cosegregations taking into account the first localisation. We found that the particular haplotype 5-6 on the chromosome 14 locus containing MYH7 showed cosegregation in the remaining affected subjects. This haplotype was found in affected subject I.3 and could have been transmitted by the dead female I.2. The SSCP screening of the first 24 MYH7 exons followed by sequencing of the abnormal profiles allowed us to identify a G to A mutation in exon 15 leading to a Glu483Lys mutation. This missense mutation was found in subjects I.3, II.2, II.3, and III.3 and also in subjects II.1 and III.5, who thus carried the two mutations (fig 1). To check that the MYH7 Glu483Lys mutation is not a polymorphism, SSCP analysis of 200 unrelated normal chromosomes was performed and failed to detect this abnormal profile. In addition, this amino acid is highly conserved in different species and isoforms.
In summary, five subjects carried the MYBPC3 mutation including three clinically unaffected members, two children and one 23 year old adult, who had normal ECG and echocardiogram. Four subjects carried the MYH7 mutation, and all were clinically affected. Two patients, a woman and her son (II.1 and III.5) aged 56 and 32 years, were double heterozygotes for the two mutations (table 1). No difference in the degree and pattern of LVH was observed between patients bearing MYH7 or MYBPC3 mutations. In contrast, the degree and extent of LVH was significantly higher in the two double heterozygotes (maximal interventricular septum thickness 28 and 32 mm) than in the other affected patients (maximal interventricular septum thickness between 15 mm and 25 mm) (table 2).
Comparison of the degree and extent of LVH assessed by the maximal interventricular wall thickness (max IVS) and the Spirito score in the double heterozygous and single heterozygous patients
Discussion
This is the first report of the presence of two mutations in two distinct genes, a missense mutation in the MYH7 gene and a nonsense mutation in the MYBPC3 gene, in a family with hypertrophic cardiomyopathy, in which two members carried both mutations. Based on the prevalence of hypertrophic cardiomopathy in the general population, which is 1/500,16 these findings should occur in fewer than 1/250 000. Until now, only two compound heterozygotes for mutations in the MYH7 gene have been described, one patient carrying both a missense and a nonsense mutation in a Japanese family17 and the other carrying two missense mutations.18 In each of these two families, one of the mutations seemed to be recessive, as it was phenotypically silent in the heterozygous carriers with an unaffected allele. However, only the compound heterozygote patients were phenotypically affected, with onset at an early age. In the present family, each of the two distinct mutations was responsible for the FHC phenotype. As the two described mutations are new ones, the number of genotyped subjects carrying a mutation is not sufficient to determine the penetrance. There was no difference in wall thickness between patients with the β-myosin heavy chain mutation and patients with the myosin binding protein C mutation, as previously observed.19 Nevertheless, the degree and extent of hypertrophy were more pronounced in the two patients bearing both mutations, although the age of onset in these patients is unknown.
The MYBPC3 Glu1096 termination codon mutation is predicted to produce a truncated protein without the C-terminal domain,6 which binds to titin and myosin. The MYH7 Glu483Lys mutation is likely to affect a protein domain involved in actin fixation. These findings are consistent with previously described mutations in which most of the MYH7 mutations are missense and most of the MYBPC3 mutations are nonsense ones.20 21 As β-myosin heavy chain and myosin binding protein C interact together in sarcomeric structure and function, we can hypothesise that the presence of quantitative or qualitative abnormalities at both levels results in an increased compensatory hypertrophy owing to additional or synergistic negative effects on the contractile function. In conclusion, clinical and genetic analysis of the present family showed that two distinct mutations responsible for a FHC phenotype may be simultaneously present in patients without being responsible for a lethal phenotype. Nevertheless, the degree and extent of left ventricular hypertrophy were more pronounced in the doubly heterozygous patients. Because of the limited number of doubly heterozygous patients in our report, future studies will be needed to confirm this finding and assess its prognostic implications.
Acknowledgments
PR, RI, LC, BM, GB, PC, MK, KS, and BH are members of the Institut Fédératif de Recherche “Physiopathologie et Génétique Cardiovasculaire” No 14. This work was supported by INSERM (Réseau de Recherche Clinique No 4R009B), the Association Française contre les Myopathies, the Fédération Française de Cardiologie, and the Délégation à la Recherche Clinique de l’Assistance Publique-Hopitaux de Paris (Crédits EMUL et IFR Physiopathologie et Génétique Cardio-vasculaire). We wish to thank Dr Jacques Beckmann from Généthon for his helpful collaboration.