Article Text

Abstract
We present two patients with the full Williams syndrome (WS) phenotype carrying a smaller deletion than typically observed. The deleted region spans from the elastin gene to marker D7S1870. This observation narrows the minimal region of deletion in WS and suggests that the syntaxin 1A and frizzled genes are not responsible for the major features of this developmental disorder and provides important insight into understanding the genotype-phenotype correlation in WS.
- Williams syndrome
- elastin
- syntaxin
- frizzled
Statistics from Altmetric.com
Williams syndrome (WS, MIM 194050 (http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dis pmim?194050) is a multisystemic neurodevelopmental disorder caused by haploinsufficiency of genes at 7q11.23. The disease is generally sporadic with an incidence of 1/20 000-1/50 000 live births,1 2 although rare familial cases of autosomal dominant transmission have been reported.3 WS is associated with dysmorphic facial features, cardiovascular disease, infantile hypercalcaemia, growth deficiency, generally mild or moderate mental retardation, and a unique cognitive profile.4 The cognitive profile is characterised by a relative strength in language and auditory rote memory in contrast to a pronounced weakness in visuospatial constructive cognition.5 More than 95% of clinically defined WS patients have a de novo deletion, homogeneous in size (1 cM), suggesting that the chromosomal breakpoints may consistently fall into a narrowly defined physical region.6 In the last few years several genes have been mapped within this interval, including the elastin gene (ELN),7-9 a novel protein kinase (LIMK-1),10the replication factor C subunit 2 (RFC2),11 thewnt receptor Drosophila frizzled homologue FZD3,12 the transcript WSCR1,13 syntaxin 1A (STX1A),14 the recently isolated GTF2I,15 as well as other ESTs and transcription units of unknown function.13 While ELN haploinsufficiency has been associated with the cardiovascular and possibly connective tissue abnormalities of WS, it does not account for the remaining clinical features of the disease.7 In particular, it is not yet clear which gene(s) is responsible for the cognitive and personality profile characteristic of WS patients.5 16
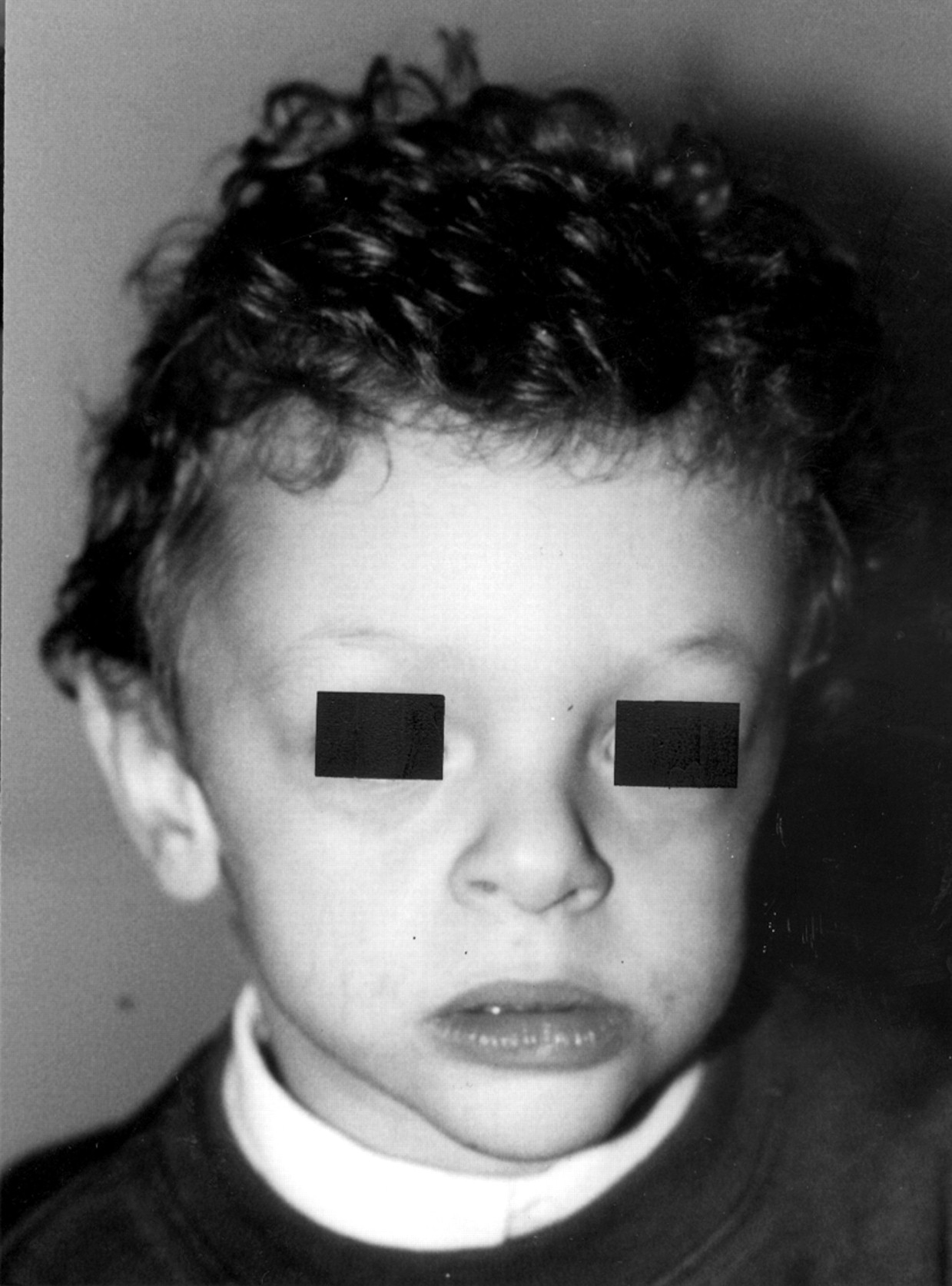
We have identified two out of 50 patients with an atypical deletion, which does not include the STX1A gene, previously shown to be constantly deleted in WS.12 14 Patient A (fig 1) was a 6 year old Italian girl who was born by caesarean section at 38 week’s gestation of an uneventful pregnancy. Birth weight was 2500 g. Apgar scores were 8 at one minute and 9 at five minutes. On examination at 6 years, the girl had a weight of 27 kg (97th centile), height of 112 cm (50th centile), and OFC of 49.5 cm (25th centile). Clinical evaluation disclosed periorbital fullness, epicanthic folds, stellate iris, malar hypoplasia, short nose with anteverted nostrils, thick lips, and bifid uvula. Echocardiography showed mild supravalvular aortic stenosis and a dysplastic pulmonary valve. Psychomotor developmental milestones were mildly retarded. She sat at the age of 11 months and walked at 20 months. Her first words were spoken at 30 months and first sentences at 5 years. The characteristic cognitive profile of Williams syndrome was noted because language abilities were relatively preserved compared to non-linguistic abilities. The child was competent in terms of lexical ability and semantic fluency. Mild articulation difficulties were present. Verbal stereotypes and social phrases were noted to be preferentially used. Difficulties in visual perceptual and visual motor abilities were recorded. The girl was hyperactive with overfriendly behaviour towards unfamiliar adults and children. Anxiety was a characteristic feature. Intelligence testing using the Wechsler Intelligence Scale for Children-Revised (WISC-R) showed an IQ of 48. She has been attending school with special assistance. Clinical records showed that serum calcium was 8.9 mg/dl at 3 days of age. Subsequent annual evaluations were also normal, ranging from 8.7 to 9.2 mg/dl.

Front view of patient A. (Photographs reproduced with permission.)
Patient B (fig 2) was a 2 year old male infant born by caesarean section at term after an uneventful gestation. Birth weight was 2300 g, length 45 cm, and OFC 32 cm. Apgar scores were 4 at one minute and 7 at five minutes. On examination, weight was 9350 g (3rd centile), length 82 cm (10th centile), and OFC 47 cm (10th centile). Physical evaluation showed sparse eyebrows, periorbital fullness, hypertelorism, a flat nasal bridge, a short nose with anteverted nostrils, and a large mouth with thick lips. Bilateral inguinal hernia was operated on at 10 months of age. Echocardiography showed stenosis of the left pulmonary artery. Psychomotor developmental milestones were mildly retarded. He sat at 8 months and walked at 18 months. At 2 years of age the infant was able to produce four to six single words and to understand many words. His behaviour was friendly, cheerful, and slightly hyperactive. Because of the child’s age, a specific cognitive profile was not detectable. Intelligence test using the Brunet-Levine Scale showed mild mental retardation (IQ=68). Serum calcium was 9.1 mg/dl at 3 months of age and 9.6 mg/dl at 2 years.

Front view of patient B.
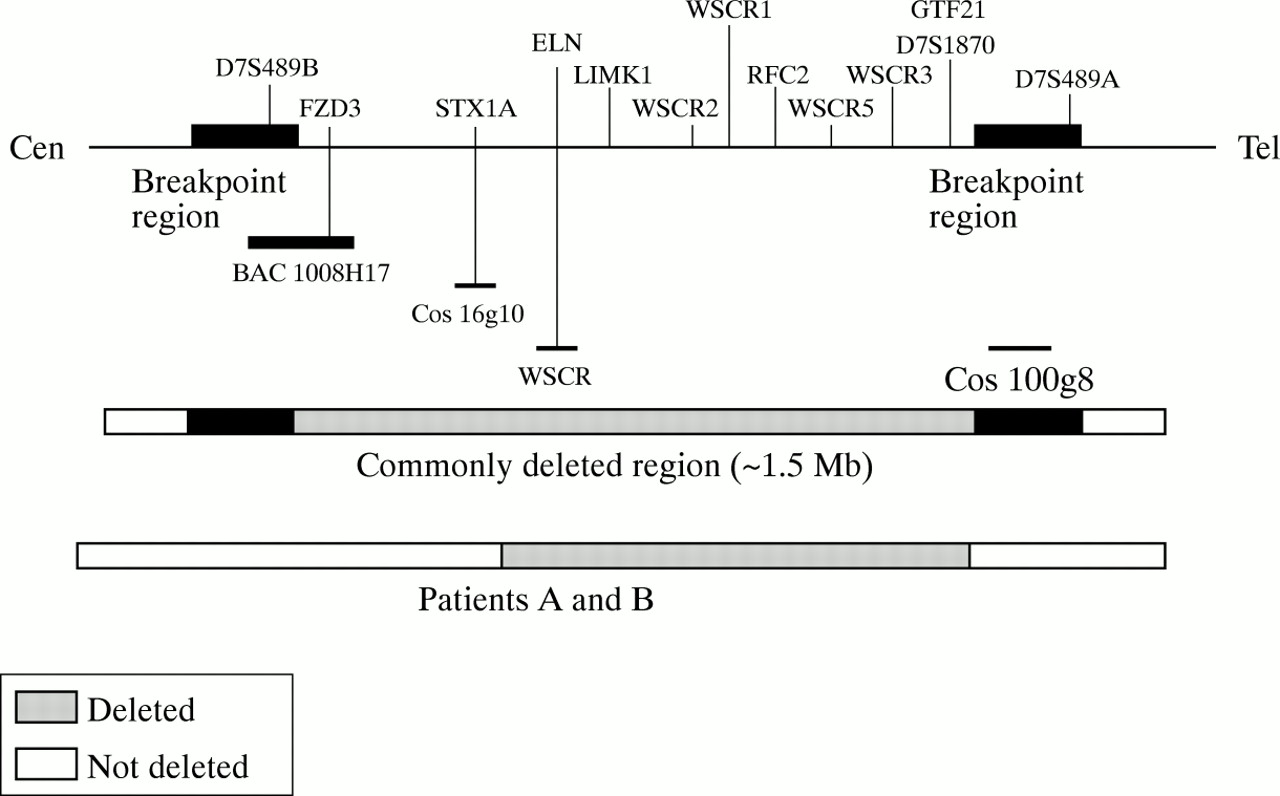
We first showed hemizygosity at the ELN locus by FISH analysis using a biotin labelled probe, WSCR (ONCOR, Gaithesburg, MD) (fig 3A). The analysis was then extended to adjacent loci and we found that the patients were dizygotic for cosmid 16g10 (STX1A gene) (fig3B).14 In order to confirm this finding a series of markers across the commonly deleted region were examined to map the boundaries of the deletion. FISH analysis showed dizygosity for BAC 1008H17 which contains the FZD3 gene and marker D7S489B, and cosmid 100g8 which contains part of the GTF2I gene and maps telomeric to marker D7S1870 (fig 4). The microsatellite marker D7S1870 detected hemizygosity and showed that the deletion was inherited maternally in patient A and paternally in patient B. These results map the extent of the deletion in both patients from between ELN and STX1A on the centromeric side, to the common breakpoint near, but not including, D7S489A on the telomeric side. This distance is estimated to be around 850 kb (fig 4).14 Further studies are in progress to refine the centromeric and telomeric breakpoints in both patients.

FISH examination of metaphase chromosomes from patient B. (A) Hemi- zygosity at the ELN locus, using the WSCR probe (red signal). (B) Dizygosity at the STX1A locus using the 16g10 cosmid probe (red signals). The telomeric probe D7S427 (yellow) was used to identify both chromosomes 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Physical map of the WS deletion region at 7q11.23 (not to scale) showing the localisation of the probes used in our FISH analysis in relation to genes and polymorphic markers (D7S489A, D7S489B, and D7S1870). The deletion analysed here does not include the STX1A gene and it is estimated to be less than 1 Mb.
This is the first report identifying typical WS patients with deletions which do not span the entire commonly deleted region. The patients described here exhibit the full spectrum of the WS phenotype, with the exception of hypercalcaemia, yet harbour 7q11.23 deletions roughly half the size of those seen in the majority of WS patients (estimated 1.5-2 Mb). To date only two genes (STX1A and FZD3) have been mapped within the region not deleted in our patients.12 14 STX1A is an integral membrane protein found almost exclusively in neurones where there is a component of the preassembled vesicle docking and vesicle fusion machinery, essential for neurotransmitter release.17 FZD3 is a novel member of a seven transmembrane domain receptor family that is the mammalian homologue of the Drosophila tissue polarity gene frizzled.12 The expression pattern and the possible role of FZD3 in wntsignalling suggest that this gene may function in different tissues during embryonic development, including the brain.12Haploinsufficiency for these two genes in WS syndrome has been postulated to contribute to various aspects of the WS phenotype, particularly the neurological symptoms.12 14 These two patients with deletions which do not include these genes, yet with a classical phenotype including mental retardation, pose additional questions in the understanding of the genotype-phenotype correlation in WS. Our observation suggests that the known genes centromeric to ELN might only be involved in hypercalcaemia and not the remainder of the WS phenotype.
These conclusions would implicate genes between ELN and the polymorphic marker D7S489A in most phenotypic aspects of WS. Genes identified so far in this region are LIMK1, RCF2I, WSCR1, GTF2I, and four partially characterised transcripts. Although LIMK1 was originally implicated in impaired visuospatial cognition,5 analysis of additional patients harbouring small deletions involving ELN and LIMK1 failed to confirm this.16 The contribution of the other genes to the WS phenotype is still undetermined. It is still possible that genes centromeric to ELN are involved in the main features of WS, either because they carry mutations not detectable by conventional FISH analysis, or because the deletions cause long range chromatin effects which influence the activity of distant genes, as proposed for the 22q11 deletion associated with DiGeorge syndrome.18 However, the deletions described here would bring the centromeric genes into an already transcriptionally active region, making this an unlikely scenario. Given the proximity of the centromeric breakpoint to STX1A, it is also possible that essential regulatory sequences necessary for the proper transcription of this gene are disrupted. Further refinement of the breakpoints in these patients will determine the contribution, if any, of STX1A to the neurological aspects of the WS phenotype.
Note added in proof
After the submission of this article, additional WS patients with atypical 7q11.23 deletions have been described by Tassabehjiet al (Am J Hum Genet1999;64:118-25).
Acknowledgments
This work was supported in part by grants from the Italian Ministry of Health.