Article Text

Abstract
Stickler syndrome is an autosomal dominant disorder with characteristic ophthalmological and orofacial features, deafness, and arthritis. Abnormalities of vitreous gel architecture are a pathognomonic feature, usually associated with high myopia which is congenital and non-progressive. There is a substantial risk of retinal detachment. Less common ophthalmological features include paravascular pigmented lattice degeneration and cataracts. Non-ocular features show great variation in expression. Children with Stickler syndrome typically have a flat midface with depressed nasal bridge, short nose, anteverted nares, and micrognathia. These features can become less pronounced with age. Midline clefting, if present, ranges in severity from a cleft of the soft palate to Pierre-Robin sequence. There is joint hypermobility which declines with age. Osteoarthritis develops typically in the third or fourth decade. Mild spondyloepiphyseal dysplasia is often apparent radiologically. Sensorineural deafness with high tone loss may be asymptomatic or mild. Occasional findings include slender extremities and long fingers. Stature and intellect are usually normal. Mitral valve prolapse was reported to be a common finding in one series but not in our experience. The majority of families with Stickler syndrome have mutations in the COL2A1 gene and show the characteristic type 1 vitreous phenotype. The remainder with the type 2 vitreous phenotype have mutations in COL11A1 or other loci yet to be identified. Mutations in COL11A2 can give rise to a syndrome with the systemic features of Stickler syndrome but no ophthalmological abnormality.
- Stickler syndrome
- collagen
- vitreous
Statistics from Altmetric.com
It is 30 years since Gunnar Stickler and colleagues1 2 published their report on hereditary arthro-ophthalmopathy and a decade since the condition we now call Stickler syndrome was reviewed in this journal.3 The purpose of this article is to provide an overview of this disorder in the light of recent advances in both clinical and molecular genetic analysis.
Clinical features
Stickler syndrome is a dominantly inherited disorder of collagen connective tissue with predominantly ophthalmic, orofacial, auditory, and articular manifestations. It is the commonest inherited cause of rhegmatogenous retinal detachment in childhood and although the systemic features are widespread, the sight threatening complications are perhaps the most conspicuous and serious manifestations.
Stickler syndrome has been subclassified into type 1 and type 2 to reflect the locus heterogeneity (OMIM Nos 108300, 184840) and this correlates with the vitreoretinal phenotype as discussed below. The systemic features are similar for both subgroups. There are no agreed diagnostic criteria for Stickler syndrome. The criteria we have used for research purposes are (1) congenital vitreous anomaly (figs 1 and2) and, in addition, any three of the following: (2) myopia with onset before 6 years of age, usually stable (fig 3),(3) rhegmatogenous retinal detachment or paravascular pigmented lattice degeneration (fig 4), (4) joint hypermobility with abnormal Beighton score, with or without radiological evidence of joint degeneration (fig 5), (5) audiometric confirmation of sensorineural hearing defect, and (6) midline clefting (fig 6).

Type 1 vitreous anomaly.

Type 2 vitreous anomaly.

(A) Facial features in a 4 year old child with type 1 Stickler syndrome. (B) Same patient aged 10 years.

Typical pigmented paravascular retinal lattice degeneration.
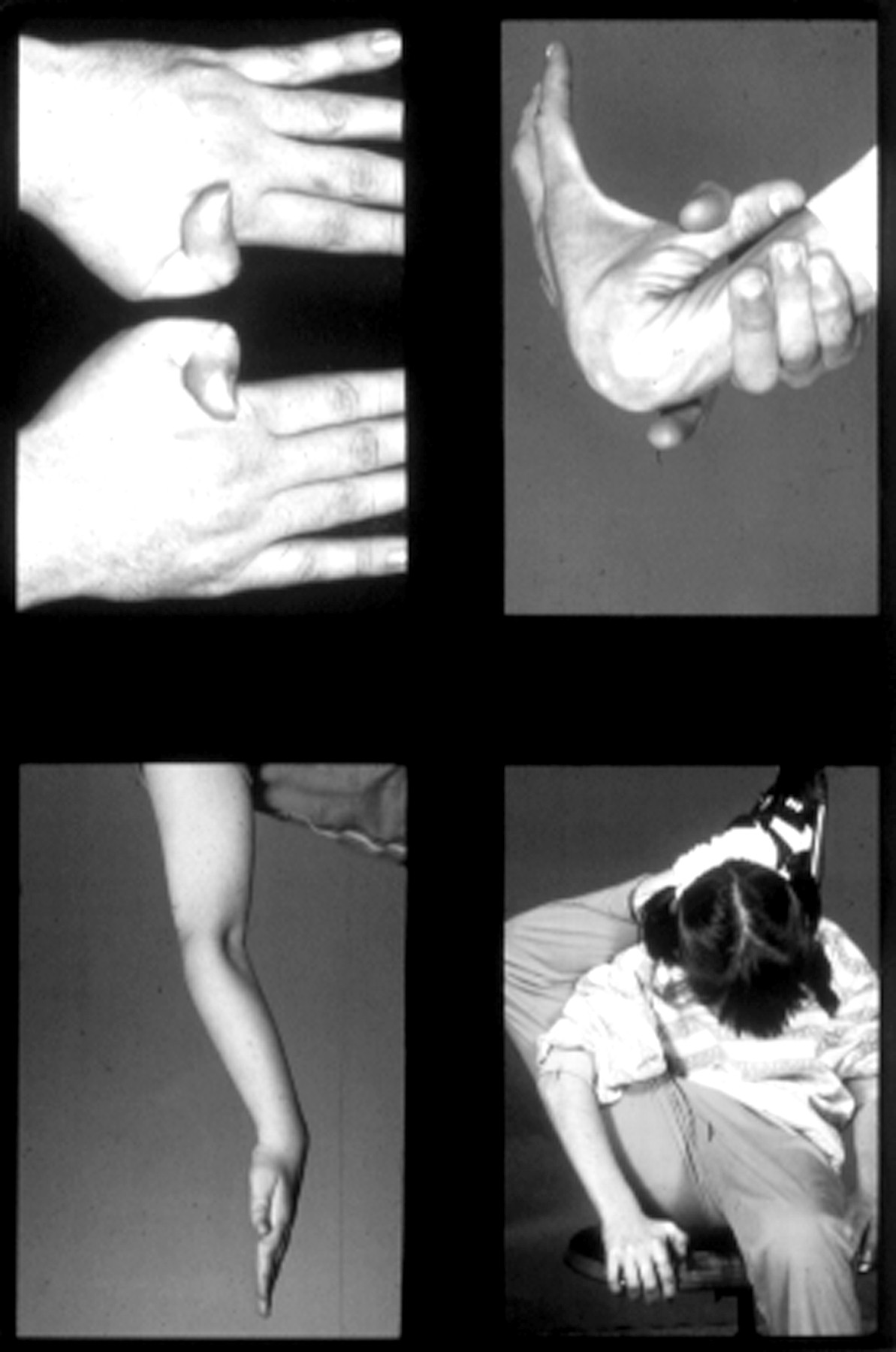
Joint hypermobility in patients with type 1 Stickler syndrome.
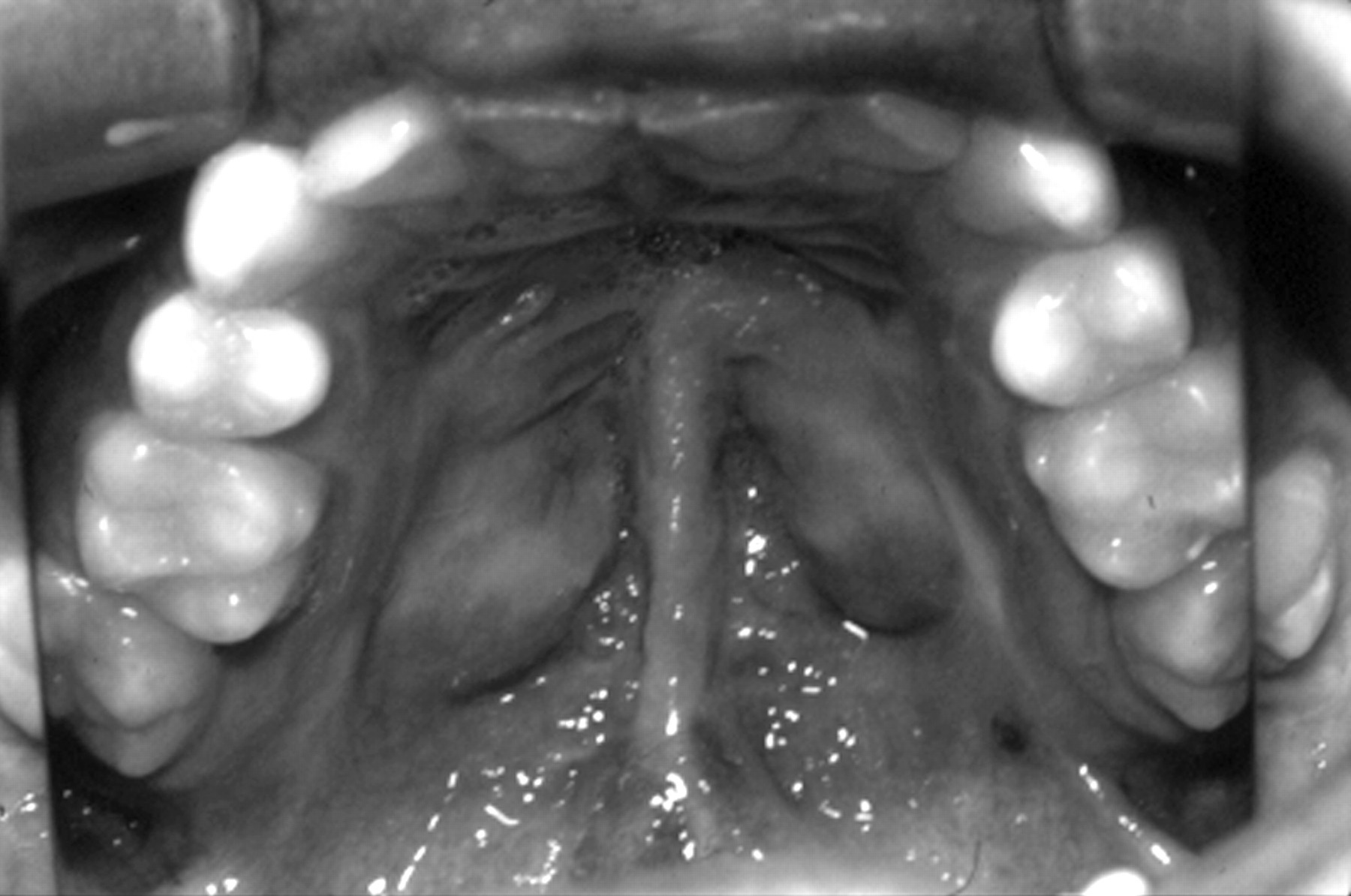
Midline cleft palate repair.
EYES
Most, but not all patients with Stickler syndrome are myopic. Unlike the common developmental type of myopia (typically with onset in the early teens), the myopia of Stickler syndrome is usually congenital, non-progressive, and of high degree. There is a well recognised association with cataract.4 5 The cataracts may be congenital and non-progressive, and many show an unusual and characteristic curved cortical distribution (fig7).

Characteristic curved cortical cataract.
Abnormalities of vitreous formation and gel architecture are pathognomonic of Stickler syndrome and in our view a prerequisite for diagnosis.6 Two distinct phenotypes can be recognised.7-9 The majority of patients have a characteristic congenital anomaly of the vitreous (the type 1 phenotype, fig 1) and this correlates with defects in type II procollagen.7 8 An apparently vestigial vitreous gel occupies the immediate retrolental space and is bordered by a distinct folded membrane. In a minority of pedigrees there is a different phenotype with sparse and irregularly thickened bundles of fibres throughout the vitreous cavity (the type 2 phenotype, fig 2).
Developmental abnormalities of the anterior chamber drainage angle predispose patients to glaucoma,10 but the most serious ophthalmic complication relates to the high risk of retinal detachment, usually as a result of giant retinal tear formation. At the time of Stickler’s original report, giant retinal tear was generally considered untreatable and blindness ensued. Modern ophthalmic surgical techniques now allow successful retinal reattachment but the risk of sudden and bilateral blindness remains a threat in patients with both vitreoretinal phenotypes.
OROFACIAL FEATURES
Classically, patients show a flat midface with a depressed nasal bridge, reduced nasal protrusion, anteverted nares, and micrognathia (fig 3A). These findings are usually most evident in childhood and with increasing age often become less distinctive (fig 3B). This is well illustrated in the previous review.3 The facial features are so variable that in isolation they are unreliable for making a diagnosis. A quarter of patients have some evidence of midline clefting. This can range from the extreme of the Pierre-Robin sequence, through clefting of the hard/soft palate, to the mildest manifestation of bifid uvula.
DEAFNESS
Patients with Stickler syndrome may suffer hearing difficulties for two reasons. Firstly, the association with cleft and highed arch palate leads to an increased incidence of serous otitis media causing a conductive hearing deficit which may be remedial. In some patients a mild conductive element persists because of ossicle defects.3 Secondly, there can be an associated sensorineural defect. Forty percent of Stickler syndrome patients show some evidence of sensorineural hearing loss, which is typically high tone and in many patients so subtle that they are unaware of the deficit.
The pathogenesis of the sensorineural hearing loss in Stickler syndrome patients is unknown. Chondrodysplastic mice with type II collagen defects show marked hearing impairment when tested with brain stem auditory responses.11 The temporal bone showed underdevelopment of the organ of Corti in the lower turn of the cochlea. In addition, there were no supporting cells, inner or outer hair cells, nerve endings, or pillar cells. The upper part of the organ of Corti, however, was almost normal in structure. These findings provide a possible explanation for the sensorineural component to the hearing loss in Stickler syndrome, particularly as this is also typically a congenital neural deafness. Whether this mirrors the facial, mandibular, and external auditory developmental delay evident in these patients is unknown (see below) and the frequency of true progression is difficult to ascertain. The cross sectional study by Lucarini et al 12 did not support a correlation between hearing loss and orofacial abnormality.
JOINT ABNORMALITIES
Many younger patients with Stickler syndrome have joint hypermobility (fig 5) and the diagnosis should be considered in hypermobile patients who are myopic. Joint mobility should be assessed objectively using the Beighton scoring system to allow comparison with an age, sex, and race matched population.13
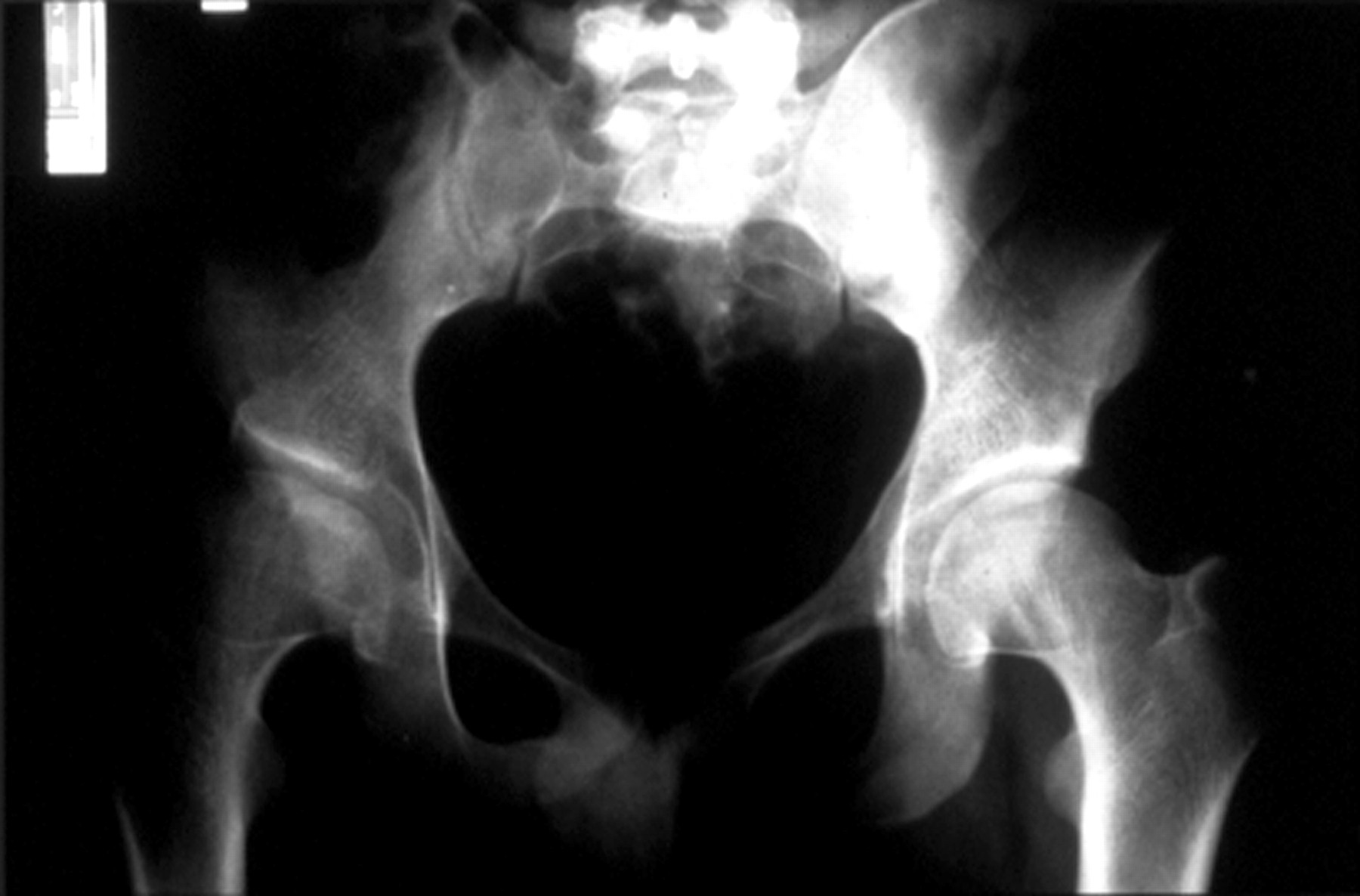
With increasing age the hypermobility reduces or is lost completely and a degenerative arthropathy of variable severity may develop by the third or fourth decade14 (fig 8). Typical radiological changes show irregularity of articular contour and loss of joint space. By middle age some patients require joint replacement surgery for hips or knees.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Degenerative arthropathy (right hip) in a 28 year old male.
OTHER FEATURES
Slender extremities, long fingers, and normal height characterise the body habitus.15 Mild spondyloepiphyseal dysplasia is often apparent radiologically. Mitral valve prolapse was found in almost half the patients in one reported series16 and, as a result, screening for valvular disease and antibiotic prophylaxis before surgery have been recommended.3 However, this is not supported by our experience of echocardiographic screening in a series of over 100 Stickler syndrome patients, none of whom showed evidence of mitral valve prolapse.9
Molecular genetics
The suggestion that disorders in connective tissue proteins,6 and specifically collagen, may underlie Stickler syndrome has been substantiated by recent research. Because of the combination of articular, auditory, and ocular abnormalities in Stickler syndrome, fibrillar collagens in particular have been leading candidates for investigation.
Collagen is the major macromolecule of most connective tissues.17 18 It consists of three polypeptide chains which are folded into a rod-like triple helical molecule. Each of the constituent chains of the triple helix are called α chains and are coiled in a left handed helix with three amino acids per turn. The constituent amino acids are regularly arranged in the order Gly-X-Y such that glycine, which is the smallest of all amino acids, occupies the restricted space in which the three α helical chains come together. This is crucial for the stability of the macromolecule.
So far, 19 collagen types have been identified designated by the Roman numerals I-XIX.19 These collagen types are formed by trimer combinations of three polypeptide chains designated by Arabic numerals. These chains may be the same or different so that the collagen molecule may depend on the products of one, two, or three genes. There are over 30 genes coding for the different types of polypeptide chains.
The repetitive sequence of collagen amino acids Gly-X-Y means that the coding sequence is also highly repetitive. Furthermore, collagen gene exons are typically multiples of nine base pairs, common standard exon sizes being 45, 54, 63, 81, 108, and 162 base pairs.20
The majority of patients (perhaps 75%) with Stickler syndrome have the characteristic type 1 vitreous phenotype and show linkage to the gene encoding type II collagen (COL2A1) on chromosome 12q13.7 8 Type II collagen is one of the group of fibrillar collagens, namely, I, II, III, V, and XI21 and is found chiefly in cartilage, vitreous, and nucleus pulposus.22 Type II collagen is a homotrimer of three α1(II) procollagen chains. Mutation screening has shown a high propensity for stop mutations and in this respect Stickler syndrome appears unique among the inherited connective tissue disorders. An interesting exception is the family reported by Balloet al 23 with an Arg704Cys substitution, in whom ocular problems and conductive deafness predominated, skeletal changes resembled a mild form of multiple epiphyseal dysplasia, and unusually all affected patients had stubby digits. Other types of mutations in the COL2A1 gene have been associated with spondyloepiphyseal dysplasia congenita, Kniest dysplasia, achondrogenesis type II, hypochondrogenesis, or premature osteoarthropathy.24
In most families, with the type 2 vitreous phenotype, linkage to COL2A1 can be excluded and other collagen and collagen associated protein candidates are currently being examined. Mutations in the gene encoding the α1 chain of type XI collagen (COL11A1) on chromosome 1p21 have so far been found in three UK families and these are, to date, the only mutations associated with the type 2 vitreous phenotype.25 26 The α2 chain of type XI collagen is not expressed in vitreous27 and mutations of its encoding gene (COL11A2) on chromosome 6p21.3 have been reported in Stickler-like syndromes which lack any ocular abnormality, as discussed below under differential diagnosis. Mutations reported in Stickler syndrome are summarised in table 1.
Mutation spectrum in Stickler syndrome
In several pedigrees with the full Stickler syndrome phenotype including vitreous abnormality, we and others28 have excluded linkage to collagens II and XI showing that there is further genetic heterogeneity still to be resolved.
Diagnosis
Clinical diagnosis using the criteria we have suggested above requires slit lamp examination of the vitreous. However, in practice, it may be difficult to obtain an adequate slit lamp vitreous examination in children under 4 years of age. Molecular genetic diagnosis is not currently available on a service basis because of the size, complexity, and number of genes involved. The diagnosis of Stickler syndrome should be considered in (1) neonates with Pierre-Robin sequence or midline cleft, (2) infants with spondyloepiphyseal dysplasia associated with myopia or deafness, (3) patients with a family history of rhegmatogenous retinal detachment, and (4) sporadic cases of retinal detachment associated with joint hypermobility, midline clefting, or deafness.
Differential diagnosis
Several disorders resembling Stickler syndrome have been described and their status as distinct entities remains controversial. Molecular genetic data are beginning to inform this debate but uncertainty remains. This will only be resolved when more genotype data become available combined with detailed descriptions of the associated ocular and non-ocular phenotype.
WAGNER SYNDROME
Wagner29 reported a large Swiss family with an autosomal dominant eye disorder resembling Stickler syndrome but without retinal detachment. Many of the families subsequently reported as Wagner syndrome have had systemic features in common with Stickler syndrome and the distinction between the two conditions has become blurred. Indeed, some authors have suggested that they are the same disorder.30 Evidence that families showing only the ocular manifestations of Wagner syndrome have a condition distinct from Stickler syndrome has come from the finding of linkage to 5q13-q14 in the original Wagner family31 and exclusion of linkage to COL2A1 in another family.32 In view of these findings, the term “Wagner-Stickler syndrome” should be abandoned. Korkkoet al 33 reported a patient with Wagner syndrome resulting from a substitution of the bulky amino acid aspartate for glycine in exon 10 of COL2A1 and postulated a possible link between the type of mutation and the Stickler or Wagner phenotypes. However, frequent retinal detachment and to a lesser extent cataract were ascribed to Wagner syndrome, whereas in the original report29 no patient suffered a retinal detachment, “cataracta complicata” was almost universal, and myopia was low in all cases. It could be argued that in the family reported by Korkkoet al 33 the phenotype more closely resembles Stickler syndrome than Wagner syndrome.
EROSIVE VITREORETINOPATHY
Brown et al 34 have described an autosomal dominant eye disorder they called erosive vitreoretinopathy with a phenotype resembling Wagner syndrome and lacking any systemic abnormalities. This condition has also been mapped to 5q13-q14 suggesting it may be an allelic variant of Wagner syndrome.31
MARSHALL SYNDROME
Marshall35 reported a large family showing autosomal dominant inheritance of cataracts, myopia, abnormal vitreous, midfacial hypoplasia, and congenital deafness. Marshall thought the phenotype might represent an incomplete form of hereditary anhidrotic ectodermal dysplasia but acknowledged that the hair was normal and evidence of hypodontia and hypohidrosis was “not strongly convincing”. Shanskeet al 36 noted from the published photographs that one of Marshall’s patients had striking hypertelorism with perhaps mild hypertelorism in others. There has been much debate about whether Marshall syndrome is a distinct entity37 and, if so, whether ectodermal dysplasia is a feature of the condition.36 Ayme and Preus38carried out cluster analysis on published reports of Marshall and Stickler syndrome patients and concluded they were different. It remains to be seen whether this issue can be resolved by molecular genetic analysis. Griffith et al 39 have reported a COL11A1 mutation in a family said to have Marshall syndrome. Shanske et al 40 suggested the family had Stickler syndrome but the authors responded that, as in the original family reported by Marshall, their patients had congenital and juvenile cataracts, fluid vitreous, hearing loss, and similar craniofacial appearance and radiology.41 As these are all recognised features of Stickler syndrome and there is no information given on vitreous phenotype, the issue remains unresolved.
WEISSENBACHER-ZWEYMULLER SYNDROME AND OSMED
Weissenbacher and Zweymuller42 described a newborn with the Pierre-Robin sequence, snub nose, proximal limb shortening, dumb bell shaped femora and humeri, and coronal vertebral clefts. The parents were healthy and unrelated. Giedion et al 43 followed up the same patient at 18 years of age. Sensorineural deafness had developed at the age of 5. By adult life limb shortening had all but resolved and height and appearance were essentially normal. There was no eye abnormality. Enlarged epiphyses were a prominent radiological feature in adolescence. Giedionet al 43 reported three other patients with the same phenotype and coined the name otospondylomegaepiphyseal dysplasia (OSMED). They concluded that Weissenbacher-Zweymuller syndrome (WZS) and OSMED were the same. Pihlajamaa et al 44 subsequently showed that the original WZS patient was heterozygous for a mutation in COL11A2. Other families showing autosomal dominant inheritance of a similar non-ocular Stickler syndrome phenotype as a result of COL11A2 mutations have been described.45-47 Van Steenselet al 48 reported three sibs of consanguineous parents who were homozygous for a COL11A2 mutation and had the OSMED phenotype. Vikkula et al 46 reported several members of a consanguineous family who were homozygous for a mutation in COL11A2. They had severe congenital sensorineural deafness, midface hypoplasia, short, upturned nose, prominent eyes, prominent supraorbital ridges, and early adult onset of severe osteoarthritis of the hips, knees, shoulders, and elbows. Adult height was slightly reduced with increased lumbar lordosis. The interphalangeal joints were prominent with short fifth metacarpals. As in all reported families with COL11A2 mutations, ophthalmological examination was normal.
OTHER DISORDERS
Other disorders with some features in common with Stickler syndrome include spondyloepiphyseal dysplasia congenita,49Kniest dysplasia,49-51 and Marfan syndrome.52
Management
Once the diagnosis of Stickler syndrome has been established, a coordinated multidisciplinary approach is desirable, comprising the following. (1) Ophthalmological assessment with refraction and correction of myopic/astigmatic error. The quality of best corrected vision may be improved with contact lens rather than spectacle correction. Many centres are now offering prophylactic retinopexy to reduce the risk of retinal detachment. Because of the risk of detachment, all patients require long term follow up and should be advised that if they see new floaters or shadows in their vision they should seek urgent ophthalmological assessment. (2) Maxillofacial assessment if midline clefting is present. (3) Hearing assessment and management of combined conductive and sensorineural deafness if present. (4) Educational assessment. Although intelligence is normal, patients of school age may face considerable educational difficulties because of combined visual and auditory impairment. Educational authorities may need to be notified of a child’s special needs. Patient support and public education has been helped substantially by the formation of the Stickler Syndrome Support Group which was founded in the UK in 1994 (The Stickler Syndrome Support Group, 27 Braycourt Avenue, Walton-on-Thames, Surrey KT12 2AZ, UK. Tel: 01932 229421). (5) Rheumatological assessment and follow up is indicated in older patients who may benefit from physiotherapy for arthropathy.
Genetic counselling
Stickler syndrome shows autosomal dominant inheritance but with wide variation in expression so that the disease status of mildly affected relatives may only become apparent on careful clinical evaluation, including slit lamp examination of the vitreous. Affected members of the family should be identified so that they can be assessed for prophylaxis against retinal detachment and be offered genetic advice. For prospective parents, variation in expression complicates counselling because of the uncertainty about severity in affected offspring. First trimester prenatal diagnosis based on the analysis of linked markers may be possible in suitable type 1 families but direct mutation analysis is not currently an option for the reasons discussed above. In taking the linkage approach, careful clinical and ophthalmological examination is essential to confirm that the typical type 1 vitreoretinal phenotype is present and also to establish the disease status of relatives used in the analysis. Moreover, if the family is not large enough to provide confirmation of linkage to COL2A1, patients need to be advised that the correlation between the type 1 phenotype and involvement of COL2A1 is only supported by a modest number of families, which limits the reliability of the result. For type 2 Stickler syndrome, the linkage based approach is complicated by the unresolved locus heterogeneity and is not a realistic option for most families. In the second trimester, the finding of features such as micrognathia or cleft palate by ultrasound scanning offers an alternative approach to prenatal diagnosis, but if these are absent it by no means excludes the diagnosis.
Acknowledgments
We thank Dr Allan Richards for helpful advice and gratefully acknowledge the support of The Iris Fund for Prevention of Blindness. We thank the reviewers for their constructive comments.