Article Text

Abstract
Little is known about the mechanism of CGG instability and the time frame of instability early in embryonic development in the fragile X syndrome. Discordant monozygotic twin brothers with the fragile X syndrome could give us insight into the time frame of the instability.
We describe monochorionic diamniotic twin brothers with the fragile X syndrome who had different CGG repeats and different mental capacities, whereas the normal mother had a premutation. The more retarded brother had a full mutation in all his cells and no FMR-1 protein expression in lymphocytes, whereas the less retarded brother had 50%/50% mosaicism for a premutation and full mutation and FMR-1 protein expression in 26% of his lymphocytes.
The differences in repeat size could have arisen either before or after the time of splitting. The time of splitting in this type of twin is around day 6-7. Given the high percentage of mosaicism, we hypothesise that the instability started before the time of splitting at day 6-7.
- fragile X syndrome
- mental retardation
- CGG repeat
- monozygotic twins
Statistics from Altmetric.com
The fragile X syndrome is a common cause of hereditary mental retardation. The phenotype is characterised by mental retardation, facial dysmorphism, and macro-orchidism.1 The FMR-1 gene, involved in the fragile X syndrome, contains a polymorphic CGG repeat within the first exon. In patients with the fragile X syndrome the number of repeats is more than 200, also known as a full mutation. This is associated with methylation of a CpG island upstream of the FMR-1 gene and downregulation of the transcription of the gene resulting in absence of the FMR-1 protein (FMRP). In normal subjects, the number of CGG units is around 30. Non-penetrant carriers of the fragile X gene have CGG numbers between 50 and 200. Only upon maternal transmission can a premutation allele expand to within the affected range of more than 200 CGGs.
A different diagnostic approach is based on direct detection of FMRP in white blood cells.2 Blood smears are made and the FMRP is visualised by an immunohistochemical reaction with specific antibodies. In controls nearly all lymphocytes are stained, whereas in fragile X patients no antibody staining is detectable. Premutations cannot be detected as these alleles synthesise FMRP in normal amounts.
The full mutation is already present in oocytes3 or very soon after fertilisation.4 Little is known about the mechanism of repeat instability nor about the time frame of repeat instability during early embryonic development. Discordant monozygotic twins may help to elucidate this issue.
Here we describe discordant monozygotic male twins, one of whom had a full mutation in all his lymphocytes and no FMRP in his lymphocytes. The other brother had a mosaic pattern of a full mutation and a premutation in his blood cells and the FMRP could be detected in normal amounts in 26% of his lymphocytes.
Furthermore, we review previously reported monozygotic twins with the fragile X syndrome and hypothesise about the period in development in which the CGG repeat is unstable.
Subjects and methods
CASE REPORT
The parents are healthy and non-consanguineous. There is no family history of mental retardation. The twins were born at 30+5 weeks after premature contractions. They had a common chorion and separate amnions. Birth weights were 1360 and 1230 g for the first born and second born, respectively. Psychomotor development was delayed: they sat at the age of 1 year, walked at 2 years, and said their first words at 2 years.
At the age of 3 years 2 months the second boy was admitted to the Department of Paediatrics to investigate the cause of his retardation. Physical examination of this boy showed a height of 98 cm (40th centile) and a head circumference of 51 cm (40th centile). The hair was thin. The eyes were large with everted lateral lower lids and epicanthic folds. There was some periorbital fullness. He had a broad nasal bridge with a broad nasal tip. The upper lip had a Cupid’s bow and the lower lip was full. The ears were normal. Fetal pads were present on the fingertips. The external genitalia were normal. His behaviour showed autistic features. All of the additional investigations were normal (MRI brain scan, chromosome analysis, metabolic investigations of blood and urine), except for the molecular studies of the fragile X gene.
The first born boy was less retarded; at the age of 3 years 6 months he spoke two word sentences and understood more than the second born twin. Both had similar physical features.
DNA ANALYSIS
Genomic DNA was isolated from blood by the salting out procedure as described by Miller et al.5 Five μg of genomic DNA was digested to completion with EcoRI and subsequently digested with the methylation sensitive enzymeEagI for at least three hours. The DNA was electrophoresed on a 0.5% agarose gel, transferred to a nylon membrane (Hybond N+, Amersham), and hybridised to the pP2 probe, which detects a fragment containing the (CGG)n and the preceding CpG island.6 After overnight hybridisation at 65°C, the filters were washed in 0.3 × SSC/0.1% SDS at 65°C and the signal was detected by autoradiography. PCR analysis was performed according to Fu et al.7
PROTEIN ANALYSIS
Blood smears were made from one drop of blood within two hours after collection. Slides were air dried and stored at room temperature. The slides were used within three weeks after collection. The FMRP was visualised by using mouse monoclonal antibodies 1A1 against FMRP followed by a second step with goat anti-mouse immunoglobulins conjugated with biotin (DAKO), according to procedures described previously.2 A total of 100 lymphocytes was examined.
Results
Southern blot analysis in the second twin showed a full mutation with a characteristic methylated smear of the expanded CGG repeat of more than 2500 units (fig 1), whereas in the first twin a mosaic pattern of an unmethylated (pre)mutation of about 190-200 CGGs as well as a methylated smear of more than 2000 repeats was found. Molecular studies using PCR and Southern blotting withPstI in the mother showed a CGG repeat in the normal range of 29 repeats and an allele in the premutation range of about 130 CGGs (data not shown).

Pedigree and results of the Southern blot analysis of EcoRI/EagI double digests of genomic DNA hybridised with the probe pP2 in the twin brothers and their mother. The numbers above the lanes correspond to the numbers in the pedigree. Fragment sizes are indicated in kb at the side of the figure. M=molecular weight marker: HindIII/PstI digested lambda DNA.
In order to test if the twins were mono- or dizygotic, DNA fingerprinting was performed (data not shown) and showed with the use of RELTYPE8 that they were monozygotic twins with a probability of 4.3 × 10-4, whereas the probability of being dizygotic was 2 × 10-8. This made the chance that they were monozygotic 21 500 times more likely.
To explain the difference in mental capacity we decided to determine the expression of the FMRP in lymphocytes.2 Antibody tests on blood smears for the detection of FMRP were performed and showed that the first twin had expression of FMRP in 26% of his lymphocytes. The more retarded second twin had no expression of the FMRP in his lymphocytes. In a control, 90% expression of FMRP was found in lymphocytes. This showed that in these two boys the expression of the FMRP is correlated with their mental capacity.
Discussion
Patients with the fragile X syndrome are mentally retarded because their FMR-1 gene is methylated and not transcribed resulting in absent FMRP expression in the cells. In about 3% of males with fragile X syndrome the methylation is not 100%.9 In these males with methylation mosaicism, the level of retardation seems to be correlated with the percentage of lymphocytes expressing the FMRP.10
It is known that males with the fragile X syndrome have a smear on the Southern blot consisting of many different sized CGG repeats originating from different lymphocytes, implying that there is somatic instability of the CGG repeat. Probably there is only a certain period early in embryonic development where this somatic instability is present and it is hypothesised that somatic instability comes to an end when the expanded allele is methylated.11 Twins might give us insight into the time period of instability, because the time of splitting is known for different types of twins. In short, three combinations can be recognised on the basis of shared amniotic/chorionic cavities. That is, both embryos have separate placentas and chorions indicating splitting time before day 3 of gestation. Our cases with a common chorion and separate amnions indicate splitting before day 6-7. Monochorionic monoamniotic twins are separated before day 15.
At least three liveborn male monozygotic twins with the fragile X syndrome have been reported. Three pairs of brothers were concordant for the degree of mental retardation.12-14 One of the male twins had a different sized full mutation.13 No information about the time of twinning was available. Of the four sister pairs,12 13 15 16 one pair was concordant for mental retardation.12 Of the other three, only one sister was mentally retarded. Two of the three pairs had a significant difference in the X inactivation pattern showing that in the normal girl the X chromosome with the full mutation was preferentially inactivated. Pedigrees of the twins described are illustrated in fig 2. In the twin brothers presented here the time of splitting is known, which enables us to speculate about the time period in development in which the CGG repeat is unstable.

Previously published monozygotic twins with fragile X syndrome. Ratio: normal unmethylated/normal unmethylated and methylated.
Malter et al 3 found that the ovaries of 16 and 17 week old full mutation fetuses contained a full mutation, as was detected in the other tissues. The mothers of these fetuses carried a premutation. They concluded that full expansion may already exist in the maternal oocyte. Moutou et al 4 compared the molecular pattern of 212 mentally retarded fragile X children of 112 premutation mothers with a simulation study, showing that if transition to a full mutation is postzygotic, a much higher proportion of mosaicism should be found, which was not found in the 212 children in their study. They proposed that this is strong (indirect) evidence that the transition of a premution to a full mutation is prezygotic.
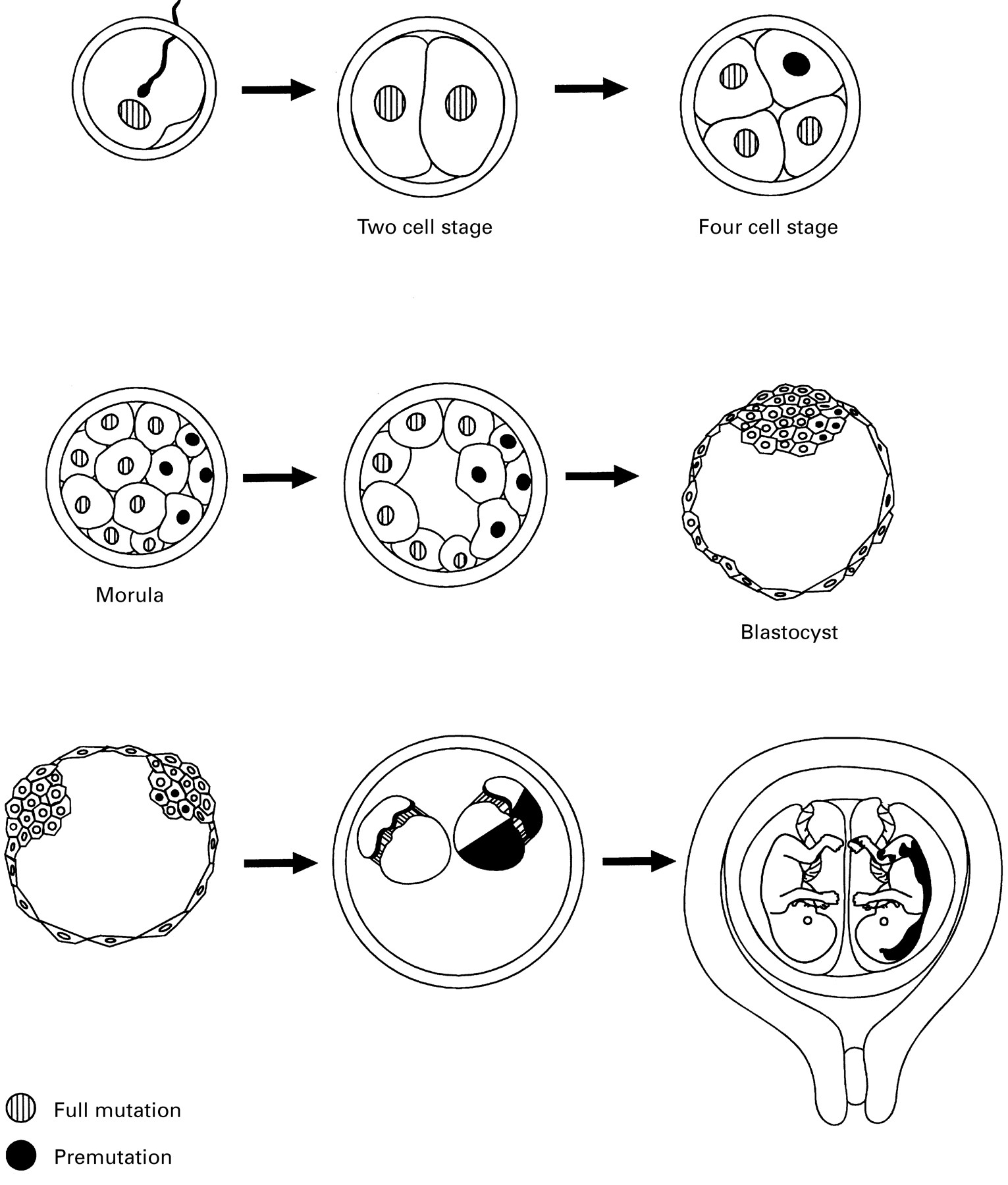
Here we describe male monozygotic twins discordant for the expression of the fragile X syndrome. The first born with mild retardation shows mosaicism for a full mutation and a premutation with 26% of lymphocytes expressing FMRP. The more severely retarded second twin shows a full mutation and the absence of the FMRP in lymphocytes. At birth, a common chorion and separate amnions were detected and therefore the splitting of the inner cell mass within the blastocyst must have been around day 6-7.17 There are two possibilities regarding the contents of the inner cell mass in these twin brothers at the time of twinning. (A) There are cells with a full mutation and cells with a premutation assuming a contraction in the early stages after fertilisation. The second born with the full mutation in all the lymphocytes developed from a cell(s) with a full mutation. The first born with the mosaic developed from a cell(s) with a premutation and a cell(s) with a full mutation (fig 3). This implies that the time of somatic instability is before day 6-7. (B) The inner cell mass consisted of only cells with a full mutation. In the second twin, all the cells developed a full mutation after splitting. In the first twin, mosaicism for a full mutation and a premutation developed after splitting, implying that in a number of cells the full mutation contracted to a premutation after day 6-7.

{kind=link}
{kind=link}
{kind=link}
Schematic representation of twinning resulting in monochorionic diamniotic twins and the instability of the FMR1 repeat as described in hypothesis A. Grey nucleus: nucleus with FMR1 full mutation; black nucleus: nucleus with premutation.
The monochorionic monoamniotic male twins described by Antinoloet al 14 were concordant for a full mutation. The time of splitting of this type (monochorionic monoamniotic) of twins is probably before day 15, so the time of expansion/instability of the CGG repeat would have to be before day 15. On the other hand, it is also possible that the concordance of the full mutation in the lymphocytes of the twins described by Antinoloet al 14 is the result of an arteriovenous anastomosis during pregnancy. In the twins described here we found a difference between the twins and therefore admixture of mature and stem cells can be excluded as playing a role in this case.
The monochorionic biamniotic male twins we describe are discordant at the molecular level, implying that the time of contraction of the CGG repeat occurred before day 6-7 if hypothesis A is true and after day 6-7 if hypothesis B is true. This is based again on the assumption that a full mutation is present in the embryo, for which evidence has been reported.3 4 Given the high percentage of mosaicism in the first twin we think hypothesis A is more likely. This study shows again that few, if any, MZ twins are genetically “identical”, because of the frequency of postzygotic mutational events, such as trinucleotide instability and imprinting at other times than gametogenetic meiosis.18
Acknowledgments
We thank the family for their cooperation. Dr P de Knijff, MGC Department of Human Genetics, is kindly acknowledged for performing the probability calculations with RELTYPE. This work was supported in part by grants from the FRAXA foundation and BIOMEDII (PL951663).