Article Text

Abstract
We describe a patient with a de novo chromosomal aberration with karyotype 46,XY,10q+, presenting clinical features of partial duplication of distal chromosome 10q. Further studies using microsatellites and FISH showed a triplication of distal chromosome 10q. The rearrangement involved both maternal homologues and the middle chromosomal 10q fragment of the triplication was inverted, similar to previously reported chromosomal triplications. Chromosomal triplications may be more frequent than assumed and may share a common molecular mechanism.
- chromosome 10q
- triplication
- FISH
- cytogenetics
Statistics from Altmetric.com
Case report
This patient is the second child of healthy, unrelated parents. He has a healthy brother and the family history is negative with regard to mental retardation or congenital malformations. Fetal movements were reduced during pregnancy. He was born at term, with a birth weight of 3800 g (50th-75th centile), length 50 cm (25th-50th centile), and head circumference 35 cm (25th-50th centile). Mild distal arthrogryposis of the hands was noted at birth. Development was delayed; he walked at the age of 20 months and spoke his first words at 5 years of age. At the age of 11 years 8 months, weight was 35.2 kg (25th-50th centile), length 153 cm (75th-97th centile), and head circumference 53.8 cm (50th-75th centile). There were minor facial anomalies, with midfacial hypoplasia, flattened forehead with a high anterior hairline, central anterior cowlick, and narrow palpebral fissures (fig 1A). Outer canthal distance was 9.3 cm (75th centile) and inner canthal distance 3.1 cm (50th-75th centile). The ears were prominent with poorly folded helices. He had hypertrophic gingiva and ventral placement of the maxillary canine teeth. A large gap was noted between the first and second toes. He was hypotonic with open mouth and reduced facial expression, hypermobile elbow joints, high, arched palate, and a lumbar scoliosis. There was distal arthrogryposis of the hands with flexion contractures at the proximal interphalangeal joints of the second to fifth fingers (fig 1B). The thumbs showed absence of flexion at the metacarpophalangeal joints. He has moderate mental retardation, with an intelligence quotient of 46 on the Terman scale, at the age of 5.5 years.
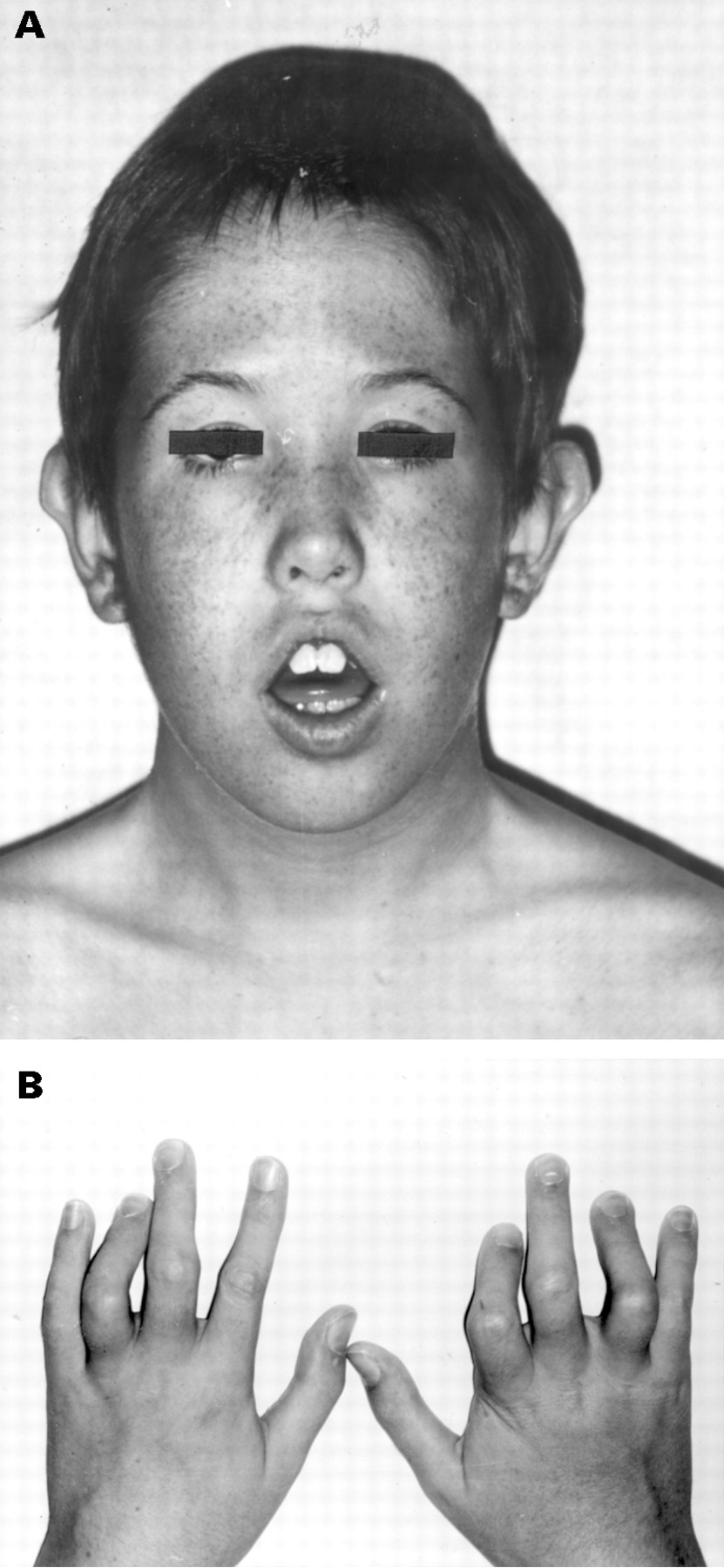
Clinical features of the patient aged 11 years. (A) Front view. Note the high forehead, hypertelorism, and hypotonia. (B) Distal arthrogryposis of the hands.
CYTOGENETIC STUDIES
The karyotype was determined on peripheral white blood cells and showed a small additional chromosomal fragment on distal chromosome 10q, with karyotype 46,XY,10q+ after high resolution G banding (fig 2). Since the clinical features were in agreement with partial trisomy of distal chromosome 10q, this fragment was tentatively identified as a partial duplication of 10q26→qter. The karyotypes of the parents were normal. An EBV cell line is available from the patient.
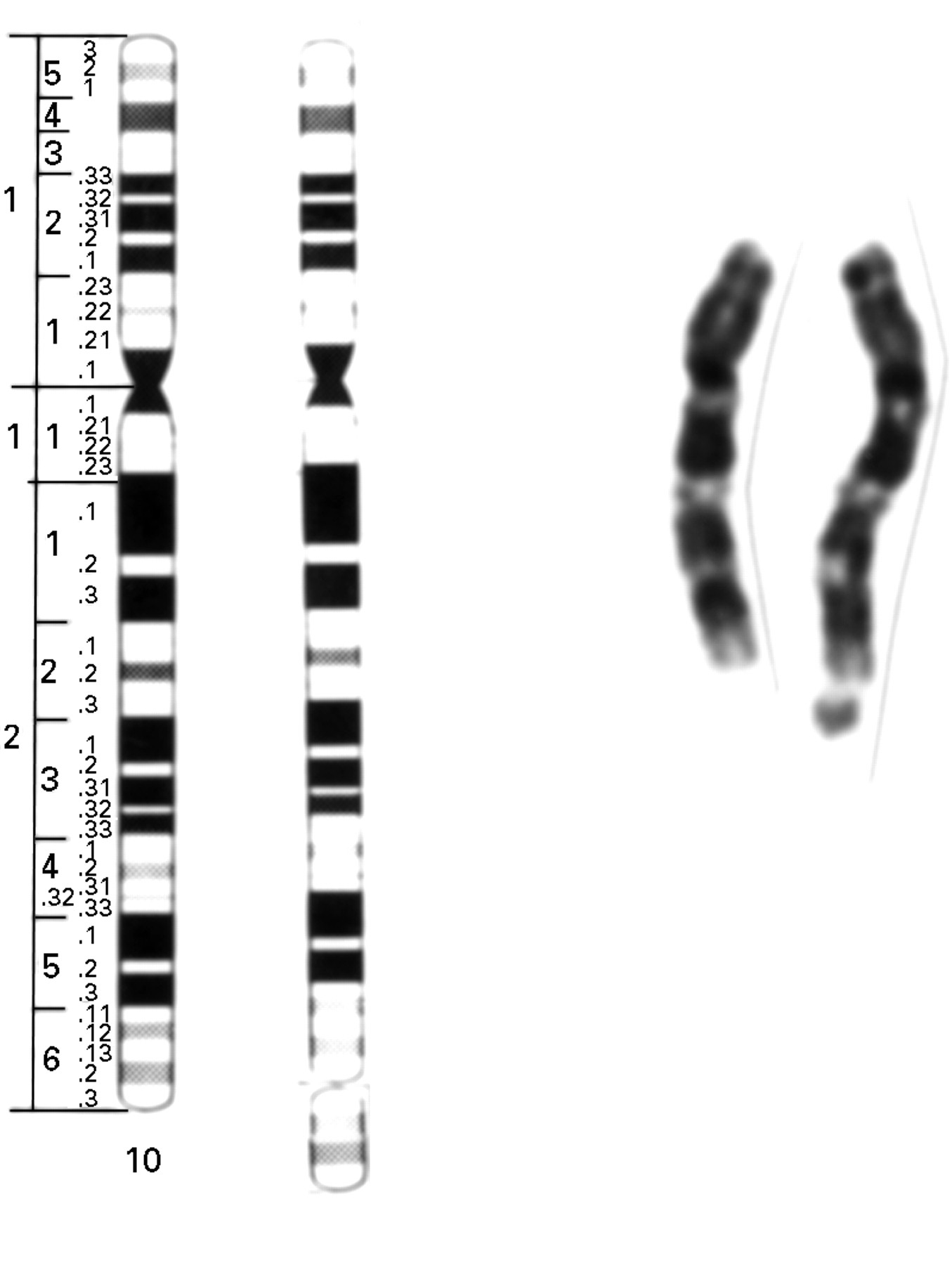
Partial karyotype showing both chromosomes 10. Note the additional chromosomal material at the distal end of one of the chromosomes 10q.
MOLECULAR ANALYSIS
A panel of microsatellite markers (CA repeats) on distal 10q was analysed on DNA extracted from white blood cells from the patient and his parents. The PCR method and analysis of the products have been described before.1 For marker D10S217, three alleles were detected, one from the father and two different alleles from the mother. However, one of the maternal alleles was present in a double dose (fig 3). This finding is compatible with a triplication of maternal origin of distal chromosome 10q. For a more proximal marker, D10S537, normal biparental inheritance was shown, indicating that the chromosomal triplication involves a fragment distal to this locus. The patient was homozygous for marker D10S198, but the mother was not, so this locus is probably not included in the triplication. The results obtained with markers D10S590 and D10S212 were also compatible with a chromosomal triplication, but the parental origin of the different alleles could not be resolved with certainty.

Analysis of microsatellite marker D10S217. Upper lane: father (homozygous). Middle lane: mother (heterozygous). Lower lane: patient. Three alleles were found, two maternal alleles (M) and one paternal allele (P). A double dose of one of the maternal alleles is present.
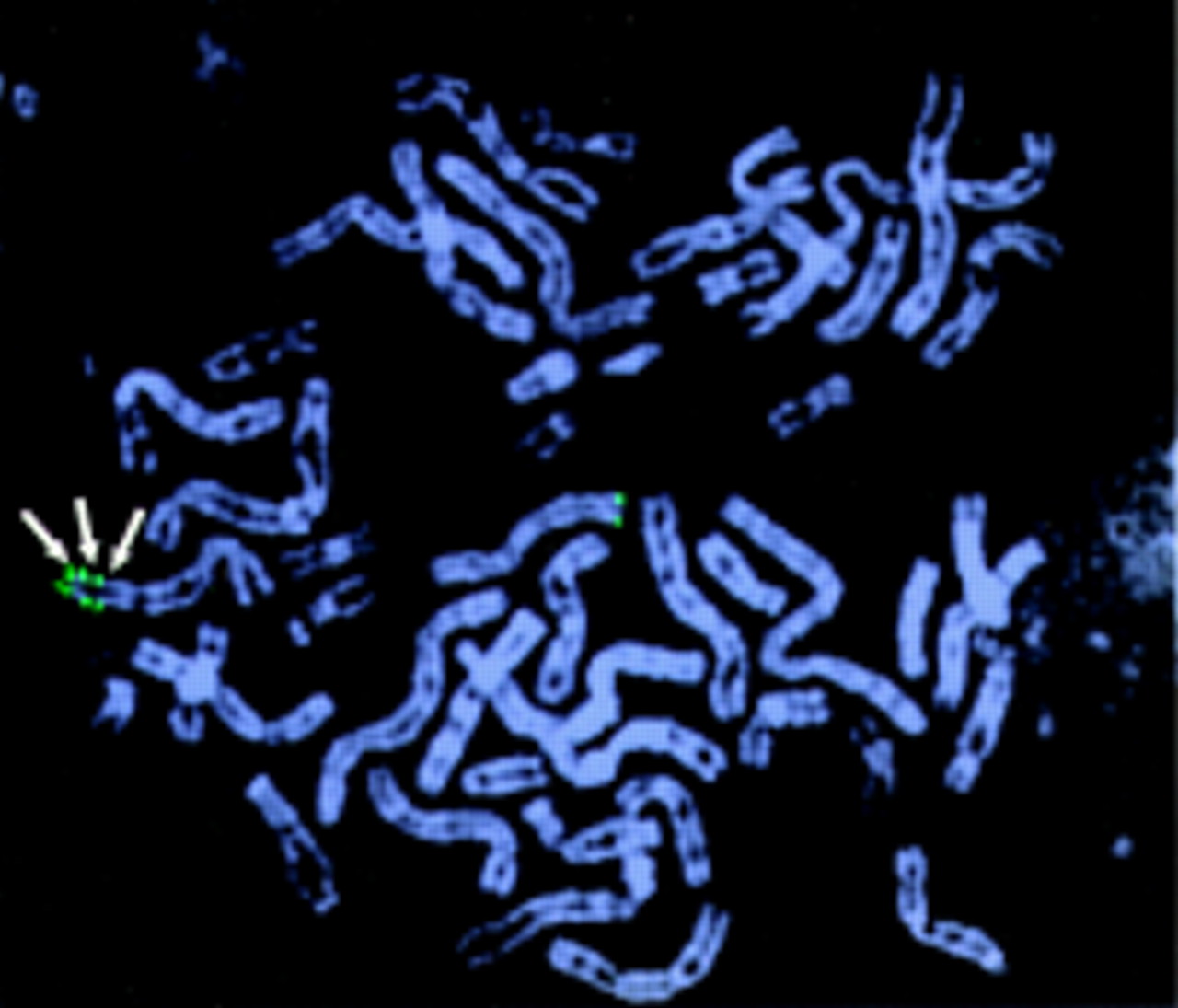
To confirm the triplication, FISH was done using probes derived from distal 10q. Three CEPH YACs were selected from the Whitehead Institute/MIT Center for Genome Research contig WC10.18, from distal chromosome 10q, which contains the marker D10S217 (Genbank accession number Z16970). With YAC 915A2, three distinct signals could be detected on the abnormal chromosome 10q, but only one signal on the normal chromosome 10q, confirming the presence of a triplication (fig4).

Triplication of distal chromosome 10q. By means of probe Y915A2, three signals on one of the chromosomes 10q were visualised (arrows), confirming the triplication of distal chromosome 10q.
To determine the precise orientation of the fragments involved in the triplication, YAC 745C10 (centromeric) and YAC 886D4 (telomeric) were labelled with a different fluorochrome. As shown in fig 5, the data support an inverted orientation of the middle fragment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inversion of the middle fragment of the triplication. (A) Schematic representation of two of the theoretically possible rearrangements resulting in a distal triplication of chromosome 10q: tandem triplication (left) or inversion of the middle fragment (right). The position of the microsatellites and YAC signals obtained by FISH are indicated. (B) FISH with probes Y745C10 (red, more proximal on 10q26) and Y886D4 (green, more distal on 10q26). The results indicate the presence of an inverted orientation of the middle fragment. (C) Subtelomeric probe of distal chromosome 10q on interphase nuclei. Note the presence of three signals in each metaphase, with two juxtaposed signals. A double intensity of one of the signals can be seen (arrow).
By means of the subtelomeric cosmid probe 2136c3 on chromosome 10q,2 a single signal was detected on the normal chromosome 10 and two on distal chromosome 10q. Interestingly, the proximal signal was consistently more intense than the telomeric signal. However, these interstitial signals never appeared clearly as two distinct signals, not even in interphase nuclei (fig 5C), which is fully compatible with the inverted orientation of the middle chromosomal fragment of the triplication. In addition, this shows that the triplicated fragment includes most of the subtelomeric region of distal chromosome 10q. FISH using a telomere specific probe1 showed no interstitial signals on the rearranged chromosome 10q (results not shown).
Discussion
In this patient with a chromosomal triplication of distal 10q, clinical findings characteristic of a duplication of distal 10q were present, including the facial features, joint contractures, and mental retardation.3 Interestingly, the manifestations in this patient were less severe than in previously described patients, who usually had major internal organ malformations (such as congenital heart defects) and more pronounced mental retardation.3 4This is surprising, since tetrasomy for a chromosomal fragment is expected to result in a more severe phenotype compared to trisomy. An obvious explanation is that in the present patient, the triplicated chromosomal fragment is smaller than the more commonly observed 10q24-qter duplication, and, apparently, the presence of four copies of genes within the distal chromosome 10q region does not further promote the genetic imbalance.
At present, only 10 patients with a constitutional chromosomal triplication have been reported, including chromosome fragments 16q12.1-q12.2,5 9p22-pter,62q37,7 5p14-p15.3,8 7p21.3-p22,9and 15q11-q13.10-13 The majority of chromosomal triplications are interstitial, with the exception of the present patient and the patient reported by Batanian et al,6 who have a triplication of a terminal chromosomal fragment. Our results indicate that the rearrangement was of maternal origin, involved both maternal homologues, and that the middle fragment was inverted. This is similar to the findings in the other patients with a triplication who were studied in detail, suggesting a similar mechanism underlying both interstitial and terminal chromosomal triplications. The precise sequence of events leading to this type of rearrangement is unclear at present, but from the finding that the two maternal chromosomes are implicated, it can be inferred that a meiosis I error is involved, possibly with an inverted duplicated chromosome 10q as an intermediate step.
The most frequently observed inverted duplication involves chromosome 8p and is associated with a deficiency of the telomeric 8p region.14 The mechanism leading to triplication seems to be different, since in the present case, as well as in the reported patient with a terminal triplication of distal 9p,6 no telomeric deletion could be shown by FISH using subtelomeric probes. However, with the currently available techniques, a very small subtelomeric deletion cannot be excluded in the present patient. Alternatively, in the patient with a terminal triplication of 9p,6 interstitial telomeric sequences were detected, suggesting the possibility that in the present patientds also short interstitial telomeric sequences might be present, undetected by current techniques.
In conclusion, the present observation suggests that patients with small, de novo chromosomal duplications should be studied for the presence of a triplication. Identification and further molecular characterisation of additional patients may lead to more insight into the mechanisms leading to these chromosomal aberrations.
Acknowledgments
The expert technical assistance of R Toelen, K Minner, and C Huysmans is acknowledged. We thank J Vermeesch for the subtelomeric 10q probe and Professor A Schinzel for helpful discussion.